CLINIQUE DES MALADIES DU MOTONEURONE
D’après
une présentation du Dr François Salachas
Centre SLA - Département des Maladies du Système Nerveux - Hôpital de la Salpêtrière
Séance du 6 Mai 2010
1. Points clés
· Dégénérescence progressive des motoneurones.
· Tableau initial distal, non systématisé, moteur pur.
· Diagnostic clinique : association de signes d’atteinte du motoneurone périphérique et central.
· Diagnostic paraclinique : EMG.
· Diagnostics différentiel : multiples, éliminer les lésions autres du système nerveux (ENMG, IRM) et les maladies mimant la SLA (ENMG, biologie).
· Prise en charge globale : précoce, un seul traitement médicamenteux étiologique, un suivi multidisciplinaire, des moyens de soulager.
2. PRINCIPALES MALADIES DU MOTONEURONE (MN)
· Les sclérose(s) Latérale(s) Amyotrophique(s) ou Maladie de Charcot (France) ; Maladie de Lou Gehrig (USA) ; Maladie du motoneurone (GB)
· La sclérose Latérale Primitive : SLP
· Les amyotrophies spinales de l’adulte
o Forme de l’adulte jeune avec délétion du gène SMN1
o Bulbo spinale liée à l’X ou syndrome de Kennedy
3. Les Sclérose(s) Latérale(s) Amyotrophique(s
3.1. Définition
C’est une affection neurodégénérative décrite par J.M. Charcot en 1869. Elle est encore parfois appelée maladie de Charcot.
Ce syndrome clinique est caractérisé par l'association d'un déficit moteur progressif central et périphérique touchant la plupart des muscles du corps, de fasciculations, d'une amyotrophie et de troubles respiratoires. Le nom « latéral » provient de la dégénérescence des faisceaux pyramidaux corticospinaux qui descendent latéralement dans la moelle épinière.
La définition anatomopathologique classique est celle d’une sclérose des cordons latéraux de la moelle épinière, des cordons pyramidaux et d’une raréfaction des MN dans la corne antérieure
3.2. Lésions
La SLA est une maladie neurodégénérative marquée par la mort sélective des neurones du cortex moteur et des moteurones du tronc cérébral et de la moelle. La perte progressive de ces deux fonctions motrices centrale et périphérique est responsable de la plupart des signes cliniques rencontrés au cours de cette affection, comme I'atteinte pyramidale, l'amyotrophie et les troubles respiratoires et bulbaires.
|
Association d’un syndrome pyramidal et d’un
syndrome neurogène périphérique |
|
|
Syndrome pyramidal |
Syndrome neurogène périphérique |
|
· Atteinte de voie pyramidale · ROT vifs, diffusés, polycinétiques · BBK, spasticité |
· Atteinte du motoneurone de corne antérieure · Déficit moteur · Amyotrophie ·
Fasciculations, crampes |
3.3. EPIDEMIOLOGIE SLA en France
La SLA a une incidence annuelle proche de 2 à 3 pour 100 000 habitants, soit 1000 nouveaux cas par an (la moitié de l’incidence de la SEP). La prévalence est de 6000 cas en France. C’est une maladie rare, mais, en France :
· Un nouveau cas toutes les 8 heures
· Un décès par SLA toutes les 12 heures
La médiane de survie est de 36 mois
L’atteinte des muscles respiratoires est constante. Dans ce contexte, la ventilation non invasive peut être proposée à 5 à 20 % des patients selon les centres.
3.4. Une groupe de pathologies hétérogène….
Le terme SLA recouvre des entités différentes. Il faudrait mieux parler des SLA…
3.4.1. Le site de début est variable
Les formes spinales
Elles sont plus fréquentes chez l’homme.
Elles débutent aux membres
L’âge moyen est situé entre 50 et 55 ans.
Les formes bulbaires
Elles sont plus fréquentes chez la femme.
Dans cette forme, les noyaux moteurs du tronc cérébral seront touchés dans leur grande majorité, à l’exception des noyaux oculomoteurs. L’atteinte des noyaux moteurs des nerfs crâniens est diffuse.
Elles débutent par une dysarthrie ou des troubles de déglutition.
L’âge moyen est plus élevé, entre 60 and 65 ans
3.4.2. Le rythme des décès est variable
Rien ne permet de prévoir précisément l'évolution de la sclérose latérale amyotrophique. Elle n'est que très rarement régulièrement progressive, laissant apparaître des périodes où la maladie semble stabilisée. Le facteur pronostic le plus important est l'atteinte bulbaire et celles des muscles respiratoires. La forme bulbaire a l'évolution la plus rapide. Certains malades décèdent en moins de 18 mois ; d’autres ont des survies supérieures à 10 ans. Le décès survient dans un délai plus ou moins rapide : 50% en 3 ans, 80% en 5 ans et 90% en 10 ans.
3.4.3. Les formes sporadiques
Les formes sporadiques représentent,
en France, 90 % des
cas. La cause initiale est inconnue et est
vraisemblablement multifactorielle associant, probablement, des facteurs
génétiques et environnementaux. Le seul marqueur spécifique de la maladie est neuropathologique et
repose sur la mise en évidence d’une dégénérescence motoneuronale associée à la
présence d’inclusions positives pour l’ubiquitine et pour la protéine TDP-43.
3.4.4. Les formes familiales
Elles sont à transmission autosomale dominante et représentent environ 10 % des cas.
Dans ces formes familiales, il existe une mutation du gène codant la superoxyde dismutase SOD1 porté par le chromosome 21. Cette mutation est présente dans 10 à 20% des cas. Dans ces formes il existe une hétérogénéité phénotypique inter- et intrafamiliale qui pourrait s’expliquer par l’existence de gènes modificateurs ou la présence de facteurs environnementaux surajouté.
3.5. Le rôle de facteurs environnementaux
Il est très discuté. Tour à tour des neurotoxines, des agents infectieux ou d’autres facteurs ont été évoqués.
Le rôle
de neurotoxines a été proposé pour expliquer les foyers de haute incidence dans
des îles du Pacifique Ouest (Guam, péninsule Kii, Nouvelle-Guinée). Si certes
des facteurs génétiques sont possibles, ils n’excluent pas des facteurs environnementaux.
Plusieurs neurotoxines ont été évoquées sans preuves convaincante.
·
L’hypothèse aluminium repose sur
o La composition du sol
o Favorisé par une alimentation pauvre en calcium
o L’observation d’une augmentation des taux d’aluminium dans le SNC
o La neurotoxicité connue de l’aluminium
· L’hypothèse Cycas circinalis repose sur l’observation des populations de l’ile de Guam. Ces populations consomment des chauves souris ; elles mêmes consommatrices de noix de cicade. Cette noix contient une excitotoxine, le BMMA qui est neurotoxique chez le singe. Cependant cette neurotoxicité est réversible et nécessite des doses très élevées
Une forme particulière sur le plan anatomopathologique
D’autres
facteurs environnementaux ont été suspectés, mais actuellement sans preuves, à
partir d’études de cas-témoins, souvent discordantes portant sur des effectifs
faibles. Ces études ont incriminé :
· Les traumatismes, l’activité sportive,
· L’exposition à des toxiques (solvant, plomb, mercure),
· L’exposition à des chocs électriques répétés
· L'hypothèse d'une origine virale de la SLA est évoquée depuis de nombreuses années en raison de la similitude de cette affection avec la poliomyélite antérieure aiguë et avec le syndrome post-polio. Plusieurs arguments ont milités en faveur d'une origine virale dans certains cas de SLA.
o Certains auteurs ont retrouvé des séquences d'entérovirus dans les motoneurones de patients avec une SLA (Berger). Ces résultats n’ont pas été confirmés par la suite.
o Des cas de SLA régressive sous traitement antirétroviral ont été décrits chez des patients infectés par le VIH (Moulignier, McGowan).
3.6. SLA Typique une caricature trop diffusée…
L’image
classique comporte la séquence d’apparition de paralysies stéréotypées depuis
les muscles des membres jusqu’aux muscles bulbaires et au diaphragme, résumée
par le concept de « marche déficitaire» en l’absence totale de troubles cognitifs, sensitifs et sphinctériens. Cette image d’une maladie irrémédiablement
mortelle par insuffisance respiratoire en 3 ans doit maintenant être nuancée.
3.7. SLA en réalité...
C’est une maladie qui se présente avec une grande diversité de présentations cliniques :
· Les formes centrales avec une bradykinésie et une raideur musculaire
· Les formes périphériques avec une amyotrophie et une faiblesse musculaire
· Les formes spinales se traduisant par une faiblesse des membres
· Les formes bulbaires associant une faiblesse des muscles de la face, de la langue et du larynx
· Les formes axiales caractérisées par une faiblesse des muscles cervicaux et des faiblesses muscles para vertébraux
· Les formes respiratoires avec un tableau de détresse respiratoire inaugurale
C’est
une maladie qui se présente avec une grande diversité de modes évolutifs :
· Les formes centrales qui aboutissent à un handicap fonctionnel sans paralysie
· Les formes périphériques qui se traduisent par un handicap fonctionnel et une paralysie
· Les formes spinales aboutissement à une dépendance rapide (MS ou marche)
· Les formes bulbaires qui se traduisent par un trouble de la communication, des pneumopathies récidivantes et la nécessité d’une gastrostomie associé à une atteinte frontale d’intensité variable
· Les formes axiales qui se présentent avec une tête tombante, une impossibilité de la station assise et debout et une atteinte profonde de la fonction diaphragmatique.
· Les formes respiratoires qui posent rapidement les indications d’une ventilation assistée voire d’une trachéotomie
3.8. La clinique
3.8.1. Dans Les formes centrales
L’examen neurologique retrouve :
· Des signes pyramidaux proches de ceux rencontrés dans la SEP
o Un Babinski/Hoffmann/
o Des ROT diffusés, polycinétiques
o Une spasticité
o Un reflexe massétérin vif comme dans les syndromes pseudo-bulbaires
· Des signes « extrapyramidaux » voisins de ceux observés dans la maladie de Parkinson
o Une rigidité plastique
o Une bradykinésie
o Un trouble prosodique (rythme parole)
3.8.2. Dans les formes périphériques
L’examen neurologique met en exergue :
· Signes d’atteinte du motoneurone périphérique comme dans l’amyotrophie spinale)
o Une amyotrophie avec fasciculations et crampes
o Des ROT faibles ou abolis
o Une hypotonie musculaire
· Souvent un tremblement d’attitude
3.8.3. Les SIGNES associés
Ils
comprennent
· Une fatigabilité musculaire*
· Une asthénie permanente
· Un amaigrissement, parfois inaugural pouvant culminer par une cachexie, d’où l’importance du suivi de l’IMC+++
· Des sueurs, dont l’origine est discutée, dysautonomie, métabolique ou respiratoire
· De la dyspnée, caractéristique surtout lorsqu’il existe un facteur positionnel, un balancement TA
· Des troubles digestifs
o TD haut avec RGO et/ou gastroparésie
o TD bas avec météorisme, constipation pouvant aggraver le dysfonctionnement respiratoire
· Des douleurs, un syndrome anxio-dépressif
3.9. FORMES CLINIQUES DE SLA
Les cinq
formes classiques sont :
·
La SLA sporadique
·
La SLA génétiquement déterminée (SOD1….)
·
La SLA associée à des signes extrapyramidaux et/ou de démence…
·
La SLA avec des anomalies biologiques de signification incertaine
· Les pseudos SLA (ALS-mimic syndrome) post polio, NMBC, hyperparathyroïdie, intoxication au plomb, formes paranéoplasiques….)
Les SLA avec démence sont le plus souvent satellite d’une forme à début bulbaire. Elles s’expriment sur un mode apathique ou au contraire par une désinhibition.
Elles
peuvent également s’intégrer dans le cadre d’une démence fronto-temporale, familiale
ou non. La coexistence d'un syndrome démentiel de type
frontotemporal et d'une SLA est estimée entre 3 et 6 % des cas. Elles modifient notablement la
prise en charge par l’équipe soignante et complexifient les rapports avec
l’entourage.
4. La SCLEROSE LATERALE PRIMITIVE
C’est
une forme rare qui représente le prototype de la forme centrale pure de SLA. Elle pourrait se révéler être un syndrome à la frontière entre
plusieurs maladies hérédo-dégénératives. Dans cette forme, le pronostic vital n’est pas mis en jeu et il n’y a
pas d’atteinte diaphragmatique significative. En revanche, elle pose des
problèmes fonctionnels, comme : le traitement de la raideur musculaire, la
prise en charge de la rééducation et/ou de l’orthophonie, ainsi que l’ergothérapie
et les appareillages à proposer.
5. Les AMYOTROPHIES SPINALES
5.1. En bref…
C’est
le prototype de l’atteinte isolée du motoneurone périphérique. Elle ne met pas
en jeu le pronostic vital, du moins pour les formes de l’adulte. La progression
de la faiblesse musculaire est assez lente bien que l’handicap à terme puisse
être non négligeable. Dans ces formes il y a un déterminisme génétique fréquent.
Le tableau ci-dessous présente les différentes formes cliniques.
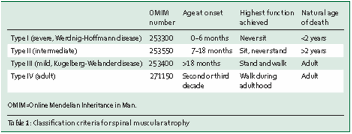
Le mode
de présentation est de type pseudo-myopathique, avec :
· Un déficit moteur proximal et une amyotrophie
· Souvent associé à une élévation des enzymes musculaires
· Une transmission génétique sur un mode récessif et l’absence d’histoire familiale.
5.2. Par délétion du gène SMN1 (maladie de Kugelberg Welander)
Forme
mineure de la maladie de Werdnig-Hoffman (voir tableau ci-dessus) qui touche
les très jeunes enfants. Elle est la conséquence d’une délétion de l’exon 7 du
gène SMN1.
Un diagnostic
moléculaire est possible depuis
1993. De ce fait, un conseil génétique est justifié par la fréquence de l’hétérozygotie
(porteur asymptomatique)
5.3. Atrophie bulbo-spinale liée à
l’X (Syndrome de Kennedy)
Cette maladie est liée à une expansion de triplets « CAG » au sein du premier exon du récepteur aux androgènes. La protéine mutée comporte une polyglutamine de longueur anormale.
La transmission est récessive liée à l’X, les femmes étant vectrices.
Le syndrome de Kennedy débute vers 35 ans en moyenne.
Les modes de présentation sont variables, dans 50 % des cas ils sont de type pseudo-SLA, dans les autres cas, ils sont de type pseudo-myopathique. Une précession fréquente par un tremblement est observée. L’existence d’une histoire familiale et d’une gynécomastie est inconstante.
Sur le plan clinique il existe des signes neurologiques et endocriniens qui sont évolutifs et des signes sensitifs infracliniques mais détectable à l’EMG. Ce tableau neurologique et endocrinien est évolutif.
Le diagnostic clinique se fait par la mise en évidence de l’association d’une fonction conservée associée à une amyotrophie qui s traduit par l’’existence de myokimies faciales associée à une atrophie linguale
Le syndrome de Kennedy ne met pas en jeu le pronostic vital car il n’ya pas de faiblesse diaphragmatique. Cependant l’handicap fonctionnel est loin d’être négligeable du fait d’une atteinte du psoas et l’existence, fréquente, de fausses routes. Ceci souligne, dans ce syndrome, l’importance de la kinésithérapie active et de l’ergothérapie.
6. RETENTISSEMENT DE LA SLA
Il est fonctionnel (handicap), musculaire, articulaire et para-articulaire, respiratoire, digestif, nutritionnel et psychique
6.1. Le BILAN FONCTIONNEL
Il
est facilité par l’utilisation d’échelles adaptées à l’évaluation du handicap
dans la SLA. Il peut être affiné par l’hospitalisation (équipe soignante…) et
permettre la prescription précoce d’appareillages et d’aides au handicap du
fait de l’évolutivité de la maladie et de la nécessité d’une anticipation
minimale des phases d’aggravation. Le tableau ci-dessous présente l’échelle
clinique.
|
ALSFRS |
4 |
3 |
2 |
1 |
0 |
|||
|
Écriture |
Normale |
Lente : mots
lisibles |
Certains mots
sont non lisibles |
Prend le stylo, n’écrit plus |
Ne peut pas prendre le stylo |
|||
|
Nourriture Sans gastrostomie |
Normale |
Lente sans aide |
Aide partielle Peut
couper |
Aide partielle ne peut pas couper |
Aide permanente |
|||
|
Nourriture Avec gastrostomie |
Normale |
Manipule de façon indépendante |
Aide intermittente |
Aide régulière |
Ne peut rien
faire seul |
|||
|
Habillage Hygiène |
Normal |
Lent avec effort |
Aide intermittente |
Aide permanente |
Ne peut rien faire seul |
|||
|
Lit Tourner, ajuster les
draps |
Normal |
Lent |
Avec difficulté |
Doit être aidé
pour finir le geste |
Doit être aidé
pour tout |
|||
|
Marche |
Normale |
Petite difficulté |
Avec assistance |
Bouge les jambes sans
déambulation |
Ne bouge pas les jambes |
|||
|
Monte les escaliers |
Normal |
Lentement |
Instable, fatigable |
Aide |
Ne peut pas monter |
|||
|
Dyspnée |
Normale |
Dyspnée d’effort à la
marche |
Dyspnée lors de l’alimentation, habillage, bain |
Dyspnée de repos |
Difficultés amenant à envisager
ventilation |
|||
|
Orthopnée |
Aucune |
Quelques difficultés
nocturnes |
Besoin de plus de deux
oreillers |
Ne peut dormir qu’assis |
Ne peut pas dormir |
|||
|
Insuffisance
respiratoire |
Aucune |
BiPAP intermittente |
BiPAP continue la nuit
|
BiPAP continue jour et
nuit |
Ventilation invasive |
|||
6.2. Le BILAN MUSCULAIRE
Il
comporte un testing musculaire et la prise en charge des différentes
manifestations.
·
Les crampes peuvent être atténuées par l’Hexaquine™, par exemple
·
Les fasciculations sont améliorées par le Mg++ et les anxiolytiques
·
Les myalgies sont contrôlées par les AINS et/ou les massages
· L’hypertonie est modérément contrôlée par le baclofène (Lioresal™) ou par les massages, le traitement des douleurs voire l’utilisation de la toxine botulique (Botox™)
· L’amyotrophie fait encore l’objet d’une quantification aléatoire et sa signification fonctionnelle et pronostique est encore inconnue
6.3. Le BILAN ARTICULAIRE
C’est
une étape importante qui impact la qualité de vie.
·
Une PASH est fréquente et est associée ou non à une
algodystrophie ; sa prise en charge comprend, les AINS, les massages,
l’utilisation de la calcitonine Cibacalcine™ s/cutanée. Il est important, chez
ces malades de casser le cercle vicieux raideur/douleur…
·
Les tendinites du moyen fessier, du biceps….
·
Les douleurs rachidiennes et para-rachidiennes (cervicales+++)
6.4. Le BILAN DIGESTIF
Le
principe est de rechercher et de traiter des troubles d’apparence banale ou
inéluctable.
·
Traiter le reflux G.O qui aggrave l’anorexie et l’hypersalivation par
le Motilium™, les antiacides
·
Contrôler la constipation, facteur aggravant l’anorexie de la
décompensation respiratoire
·
Traiter rapidement toute aggravation récente et éviter situations
extrêmes souvent préjudiciables sur un terrain fragile : fécalomes,
diarrhée sous Rilutek™, gastro-entérite…………….
·
Être attentif aux phénomènes de ballonnements (gastrique et/ou
intestinal) car ils sont fréquents et qu’ils peuvent avoir des conséquences
parfois dramatiques si état respiratoire est précaire
6.5. Le BILAN NUTRITIONNEL
6.5.1. Le contexte
Du fait d'une anorexie plurifactorielle, de troubles de déglutition et de salivation, de la dénervation et d'un hypermétabolisme, une dénutrition est présente dans 15 à 30 % des cas.
Il est essentiellement quantitatif, poids et IMC. Les objectifs sont :
· Le maintien d’un poids stable
· La prévention des troubles digestifs
Les moyens sont comprennent :
· L’optimisation de la déglutition (ortho+++)
· Le recours aux suppléments caloriques (+/- orexigènes) mais pas de régime hypocalorique
· La proposition d’une gastrostomie ou de PAC, en cas d’échec
6.5.2. LA GASTROSTOMIE
Il n’y aucun consensus sur les indications. En général, l’indication est posée sur
· Une hydratation impossible
· Une perte de poids excédant 10 à 20%
· Des repas trop longs et fatigants (bulbaires)
Il
faut savoir qu’elle est d’autant mieux supportée qu’elle est précoce. Elle est
mise en place par voie percutanée, le plus souvent. Elle autorise une recharge
calorique lente sous traitement prokinétique (érythromycine) et permet un retour
à domicile rapide.
6.6. Le BILAN PSYCHIQUE
Il est conditionné par l’évolutivité de la maladie et par l’attitude du patient vis-à-vis de son handicap, de la perception de sa place dans sa famille, la société.... et de l’existence d’une qualité de vie jugée acceptable.
Il y a un intérêt certain d’un soutien psychothérapeutique précoce du patient, de son entourage. Le recours aux psychotropes est, dans la majorité des cas, possible.
Dans ce contexte, il ne pas méconnaître l’existence d’un syndrome pseudo-bulbaire responsable d’une hyper expression des affects qui peut être contrôlé par un antidépresseur, comme le Laroxyl™.
Il faut bien jauger la qualité de la relation avec l’entourage, en cas de syndrome frontal à minima ou d’un syndrome pseudo-bulbaire.
6.7. SYMPTÔMES FRÉQUENTS
L’hypersalivation
est handicapante et peut être améliorée par :
· Un anticholinergiques (P.O)
· La scopolamine en patch ou, mieux, en s/c)
· En traitant le R.G.O
Les glaires peuvent être diminuées par :
· Des aérosols, l’Avlocardyl™
· Une aide à la toux+++
· Des techniques de désencombrement (« cough assist »)
Les mycoses, très fréquentes sont redevables d’un traitement local (Fungizone™, Loramyc™).
6.8. ATTEINTE RESPIRATOIRE
6.8.1. Le contexte
C’est
plutôt l’apanage des formes évolutives des maladies du motoneurone que ce soit
la SLA ou les rares amyotrophies spinales. Elle conditionne le pronostic et nécessite
une évaluation régulière (pronostic). Elle est évaluable en clinique
quotidienne.
La
prise en charge a bénéficié ces dernières années de l’amélioration des
techniques palliatives, non invasives ou VNI.
Il
est important d’anticiper la survenue d’une défaillance de la fonction
respiratoire et de différencier les aggravations « attendues » des
évènements intercurrents (EP, pneumopathie, OAP..).
Pour
ce faire, il faut disposer d’une « Histoire Naturelle de la Maladie »
d’un point de vue respiratoire et pouvoir mettre en place, au moment opportun, une
prise en charge optimisée :
·
Par l’utilisation de la ventilation assistée non invasive (VNI) dont
les indications s’étendent
·
De mesures palliatives, en face d’une insuffisance respiratoire
terminale
6.8.2. Les moyens
Elle
peut se faire, de préférence, au lit du malade, avec ou sans instruments au
sein de centres spécialisés ou, à défaut, en suivant leurs procédures. Un suivi
longitudinal indispensable pour apprécier
·
La pente évolutive de la maladie
·
L’imminence d’une décompensation
Le rythme
du suivi sera alors calqué sur la vitesse évolutive de la SLA.
Au
cours d’une brève hospitalisation, le bilan habituel comprend une évaluation
clinique de la performance respiratoire diurne et nocturne et un bilan initial
qui sera réévalué.
Comporte
une phase purement clinique
Le
bilan paraclinique comprend au minimum la mesure de la CV, du GDS et une
oxymétrie nocturne. Il peut comporter la mesure des pressions inspiratoires
(PiMax), de la force de reniflement (SNIP) et le débit expiratoire de toux.
L’interrogatoire
recherchera des
·
Signes de gravité immédiate, céphalées matinales, cauchemars, réveils
brusques, cyanose/sueurs/tachypnée/bradypnée/tachycardie
·
Des signes d’insuffisance diaphragmatique : respiration
paradoxale, pouls inspiratoire, orthopnée, un sniff test diminué
·
Des signes d’insuffisance muscles expiratoires, toux, encombrement
laryngé
L’examen
clinique respiratoire est beaucoup plus fiable pour apprécier l’existence d’un
pouls inspiratoire, la force de la toux et la variation de la mécanique
ventilatoire lors du passage en décubitus.
6.8.3. MESURE DE LA CVL
Elle
est réalisée sur un patient en position: assise et couchée. L’interphase est
constituée par un embout simple, avec adaptateur buccal, au mieux par un masque.
Elle nécessite de la part de l’équipe soignante, pneumologue, neurologue,
infirmière, kinésithérapeute, une phase d’entrainement pour optimiser la
fiabilité de la mesure
6.8.4. OXYMETRIE NOCTURNE
C’est
une technique d’évaluation de plus en plus utilisée. Elle explore une période
de vulnérabilité respiratoire plus importante en raison du risque d’apnées
(obstacle ou centrales et de la défaillance des muscles respiratoires
accessoires lors du sommeil paradoxal.
Les
valeurs seuils encore mal définies. La valeur prédictive du décès reste assez
difficile à évaluer actuellement, en particulier la pertinence des valeurs, la
médiane?, la SpO2 minimale?, la notion de désaturation prolongée?, le
pourcentage de temps passé en dessous de 90%? 88%?
6.8.5. VENTILATION ASSISTEE non
invasive (VNI)
Le
contexte
Elle
est amenée une transformation considérable des pratiques en permettant à la
fois une amélioration de la qualité et de la quantité de vie, du sommeil et des
performances cognitive. C’est un des seuls moments de la maladie ou on peut observer
une pause, voire une amélioration.
Les indications
Symptômes
pouvant être liés à l’hypoventilation alvéolaire diurne ou nocturne associés à
des critères objectifs, comme :
·
PaCO2 > 45 mm Hg,
·
CVF < 50% de la valeur théorique,
·
SpO2 < 90% pendant plus de 5 % du temps d’enregistrement nocturne,
·
PImax et SNIP < 60% de la valeur prédite
·
Suite de décompensations aiguës
Modalités
·
Mode volumétrique ou barométrique
·
Deuxième ventilateur si dépendance > 12H/24
·
Interface adaptée, voire plusieurs
·
CPAP non recommandée
Les problèmes
non résolus
·
Quand ?
·
Suppléance ou substitution totale de la fonction respiratoire ?
·
Comment gérer la phase d’échappement (domicile, urgences…) ?
·
La décision de pratiquer une trachéotomie
6.8.6. La trachéotomie
Elle
fait souvent suite à une décompensation
aiguë chez un patient insevrable. Elle concerne une grande
proportion des malades. Elle peut prolonger la survie de plusieurs années chez certains malades. Elle
permet le contrôle des fausses routes et les aspirations trachéales. A
l’opposé, elle réduit la faculté de communiquer
du patient et impacte nettement sa qualité de vie.
Elle est souvent une difficulté supplémentaire
rendant le de maintien à domicile difficile et sous-entend une implication forte
de l’entourage.
6.8.7. En pratique…
Surveiller
et pallier la déficience respiratoire est un objectif incontournable au cours
de la SLA.
La possibilité
de contrôler la défaillance diaphragmatique dans les années à venir devrait
constituer un progrès majeur de la prise en charge de la maladie par la
combinaison de la VNI intermittente, de la stimulation électrique du diaphragme
et, peut-être, par l’utilisation de facteurs de croissance à tropisme diaphragmatique
7. CONCLUSION
Une hétérogénéité clinique de la maladie souvent méconnue
L’importance dans la flexibilité de la prise en charge clinique et psychologique
La prédictibilité très imparfaite, y compris sur le plan respiratoire
La nécessité d’optimiser l’accompagnement des patients par une meilleure approximation du pronostic fonctionnel et vital.
Un
seul médicament, le Rilutek™ (50 mg matin et soir) améliore le pronostic, prolonge
phase d’autonomie
Le
changement de paradigme, le passage d’une maladie mortelle à une maladie
chronique
8. Critères diagnostiques de la SLA
(critères révisés El Escorial)
SLA certaine
Signes
d'atteinte du NMC et du MNP dans la région bulbaire et dans 2 régions spinales
SLA probable
Signes
d'atteinte du MNC et du NMP dans au moins 2 territoires
SLA probable sur
critères paracliniques
Signes
d'atteinte du NMC dans au moins 1 territoire et atteinte du MNP visible à l'EMG
dans au moins 2 territoires
SLA possible
Signes
d'atteinte du NMC et du MNP dans un seul territoire,
Ou signes d'atteinte du NMC dans au moins
2 territoires,
Ou signes d'atteinte du MNP dans 1
territoire et signes d'atteinte du NMC dans les territoires sous-jacents
NMC = neurone moteur central ; MNP
= motoneurone périphérique ; EMG = électromyogramme