|
EPU95 – Montmorency
Neurologie
 Mise à jour du 24 Avril 2007 Mise à jour du 24 Avril 2007

Les neuropathies
Expert Pr. Alain Créange
Neurologue – CHU Henri Mondor Créteil
Séance du 3
novembre 2005
1. Introduction
Il est important, au début de cette présentation de
définir les différents cadres nosologiques dans lesquels s’inscrivent les
différentes neuropathies.
1.1. Les polyneuropathies
Ce terme est à préférer à celui de
« polynévrite » qui présuppose une inflammation. C’est une atteinte du système nerveux à
partir de l'émergence des nerfs rachidiens constitués par
la réunion des racines antérieures et postérieures à la sortie du fourreau
dural. L'atteinte des racines rachidiennes et de la corne antérieure en est
exclue. Les signes sont
« longueur dépendants »
1.2. Les neuronopathies
C’est une atteinte
isolée symétrique ou non des ganglions rachidiens. C’est donc une
atteinte sensitive pure.
1.3. Les polyradiculonévrites
C’est une
atteinte des racines nerveuses sensitives et motrices. Elle peut être
aiguë ou chronique. Par définition, elle implique une atteinte diffuse de
la totalité des fibres nerveuses, y compris les racines.
1.4. Le mononeuropathies isolées ou multiples
Ce sont des atteintes, asymétrique, des troncs nerveux.
Les troubles sont distaux, sensitifs et moteurs en général.
4
Une atteinte unique : tronculaire, radiculaire
è
radiculopathie
4
Une atteinte multiple : tronculaire è
mononeuropathies multiples,
4
Les atteintes plexiques, à part
2. Rappels
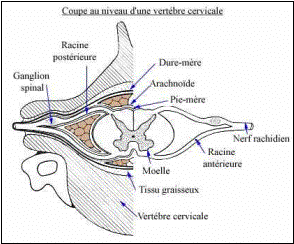 Le système nerveux
périphérique est composé de plusieurs types de fibres nerveuses. Le système nerveux
périphérique est composé de plusieurs types de fibres nerveuses.
2.1. Les fibres nerveuses
2.1.1. Fibres motrices efférentes
Le corps cellulaire est situé dans la corne antérieure
de la moelle épinière. L’axone quitte la moelle par la racine antérieure et
chemine jusqu’au muscle strié squelettique où il tient sous sa dépendance
une centaine de fibres (unité motrice).
2.1.2. Fibres sensitives afférentes
Le corps cellulaire est situé dans le ganglion rachidien
de la racine postérieure. Leur destinée médullaire les oppose en deux
groupes :
4
Lemniscales
dont les fibres cheminent dans les cordons postérieurs homolatéraux et
véhiculent la sensibilité épicritique et le sens proprioceptif
4
Spino-thalamiques
dont les fibres, qui décussent au niveau du métamère médullaire, véhiculent
de façon controlatérale le tact grossier et la thermoalgie.
2.1.3. Fibres végétatives
Ce sont les voies efférentes sympathiques et
parasympathiques. Le corps cellulaire du neurone préganglionnaire est situé
dans le tronc cérébral ou la moelle, l'axone quitte le système nerveux
central par le trajet des nerfs crâniens (III, VII, IX, X) ou des racines
antérieures médullaires pour faire un relais avec le neurone
post-ganglionnaire innervant muscles lisses et glandes.
2.2. Deux types de fibres nerveuses
Elles sont constituées d'axones (cellules nerveuses), de
cellules de Schwann (myéline) et de tissus de soutien (vaisseaux et
conjonctif).
2.2.1. La fibre nerveuse périphérique myélinisée
Elle est constituée par un seul axone engainé par une
cellule de Schwann. L'axone est formé d'une membrane plasmique (axolemme)
qui renferme l'axoplasme constitué de neurofilaments, de neurotubules, de
citernes de réticulum endoplasmique et de mitochondries. La cellule de
Schwann enserre l'axone, présente un noyau ellipsoïdal et possède de
nombreuses mitochondries. La gaine de myéline s'interrompt périodiquement
au niveau du noeud de Ranvier. La fibre nerveuse est entourée d'une gaine
fibreuse (gaine endoneurale) formée par la lame basale de la cellule de
Schwann et par un réseau de microfibrilles réticulées et collagènes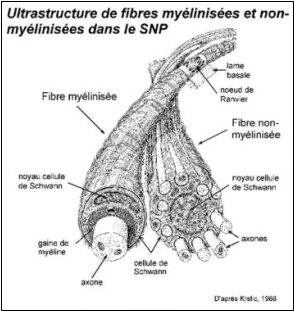
2.2.2. La fibre nerveuse non myélinisée
Elle est constituée par un faisceau d'axones associés à
une même cellule de Schwann. Chaque axone est logé dans une invagination de
la cellule de Schwann. Les fibres amyéliniques possèdent également une
gaine endoneurale faite d'une lame basale et de microfibrilles collagènes
et réticulées entrelacées
2.2.3. Les lésions élémentaires des fibres nerveuses
Ce sont :
- La dégénérescence wallérienne qui est une
désintégration progressive myélino-axonale. A cette dégénérescence
succèdent des bouquets de régénérescence axonale ou « clusters ».
- La démyélinisation segmentaire qui est une « mise
à nu » progressive des axones par destruction de la myéline au niveau
des nœuds de Ranvier qui s'élargissent anormalement.
Les lésions s'organisent en :
4
Atteintes primitives des fibres nerveuses (axone ou
myéline)
4
N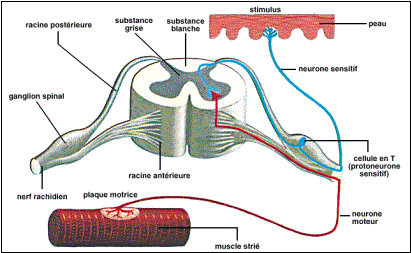
europathies interstitielles (amylose) et des structures
conjonctivo-vasculaires,
4
Neuropathies vasculaires (PAN).
3. L’approche
diagnostique
Lors de la séance, elle a été faite à partir de 4 cas
cliniques. Pour le compte-rendu, une approche « classique » a été
retenue.
Dans tous les cas, elle s'appuie sur des éléments
cliniques
- La
topographie des signes : uni ou bilatéraux, symétriques ou non
- Le
mode d'installation du tableau
- Le
contexte : diabète, médicaments, traumatisme,...
Les éléments paracliniques comprennent, principalement,
l’électromyogramme et des examens biologiques simples. Il faut souligner
que le scanner ou l’IRM cérébrale, ne sont pas des examens à réaliser pour
l’exploration des neuropathies périphériques.
4. Les
polyneuropathies
4.1. Généralités
Elles sont « longueur-dépendante. » Elles
résultent d'une atteinte diffuse et symétrique intéressant les extrémités
distales des membres, s'opposent aux mononeuropathies multiples, liée à une
atteinte successive dans le temps et l'espace de plusieurs troncs nerveux
et impliquant un processus physiopathologique différent.
4.2. Le bilan
4.2.1. Cliniquement
Les troubles débutent à l'extrémité distale des membres
inférieurs dans les formes habituelles sensitivo-motrices.
4
Troubles
sensitifs : ils sont souvent initiaux, touchant les extrémités des
membres inférieurs (paresthésies permanentes, dysesthésies, brûlures).
L’atteinte des grosses fibres myélinisées se traduit par des troubles de la
sensibilité profonde (ataxie).
4
Troubles
moteurs : au début le patient rapporte des difficultés à la
marche, une fatigabilité anormale. Puis s’installe un steppage (déficit
releveurs du pied), ensuite le déficit progresse pour intéresser la racine
puis les membres supérieurs. Il épargne les muscles respiratoires et les
nerfs crâniens. Il existe parfois des crampes (mollets, plante des pieds).
4
Troubles
végétatifs : ils sont liés à une atteinte des fibres amyéliniques
o
Hypotension artérielle orthostatique,
o
Troubles vésico sphinctériens,
o
Dysfonction érectile,
o
Troubles digestifs (diarrhée, constipation).
4.2.2. A l’examen…
Les signes suivants sont plus ou moins complètement
associés.
4
Des signes sensitifs, souvent les premiers à
apparaître :
o
Signes subjectifs :ces sensations anormales
sont des paresthésies (picotements, fourmillements, engourdissements
spontanés), des dysesthésies (déclenchées par le tact) ou des douleurs
(brûlures, décharges électriques, striction). Leur origine peut être
radiculaire ou tronculaire.
o
Les signes objectifs sont en revanche absents ou
discrets.
o
L’atteinte objective, rarement dissociée : les
troubles concernent les sensibilités superficielle [au tact (épicritique),
à la température et à la piqûre (thermo algique)] et proprioceptive
[altération du sens vibratoire, du sens de position des segments de membre,
avec signe de Romberg].
4
Des signes moteurs
o
Une paralysie ou parésie, par atteinte de la
motricité volontaire, réflexe et automatique par souffrance de la voie
finale commune (motoneurone alpha). Plus ou moins précédée d'une
fatigabilité, elle est complète (car affectant tous les types de
motricité), flasque et hypotonique.
o
Une amyotrophie retardée de trois semaines par
rapport à la lésion nerveuse.
o
Des fasciculations, spontanées ou provoquées par la
percussion ou l'exposition au froid, traduisant l'activité spontanée d'une
unité motrice. Elles sont en faveur d'une lésion proche de la corne
antérieure (motoneurone, racine).
o
Une diminution ou une abolition des réflexes
ostéotendineux.
L’examen confirme l’atteinte
symétrique, à prédominance distale, des membres inférieurs
4
Une abolition des réflexes achilléens
4
Un déficit moteur affectant les releveurs du pied
4
Une amyotrophie
4
Un déficit sensitif qui peut être discret (examen
particulièrement attentif)
4
Une atteinte végétative (peau et phanères,
hypotension).
4.2.3. Les examens complémentaires
L’électromyogramme est indispensable dans l'enquête
étiologique en permettant de mettre en évidence le processus en cause,
axonal ou démyélinisant.
Le bilan sanguin de départ est simple mais
systématique : NFS, CRP, glycémie à jeun et post-prandiale (ou mieux 2
heures après la prise de 75 g oral de glucose à la recherche d’une
intolérance au glucose), recherche d’une immunoglobuline monoclonale par
une immunoélectrophorèse des protéines plasmatiques avec dosage pondéral
des immunoglobulines, une sérologie VIH voire VHC.
4.3. Les étiologies
La recherche d’une étiologie doit prendre en compte le
type de neuropathie et son mécanisme. Les aspects suivants peuvent entrer
en ligne de compte.
4
L’âge
o
Enfant (rare et héréditaire)
o
Adulte (près de 200 causes)
o
vieillard (recherche souvent infructueuse)
4
L’origine – Ethnie
o
Lèpre (neuropathie plus fréquente dans le monde)
o
Amylose portugaise
4
Le caractère familial, surtout dans les
neuropathies chroniques
4
Les circonstances de diagnostic :
o
Maladie générale (diabète, insuffisance rénale),
o
Prise de médicaments,
o
Piqûres d'insectes.
4
Le mode d'installation : qui reste déterminant :
aigu, subaigu ou chronique.
!4.3.1 Le diabète
C’est une des causes les plus fréquentes. L'incidence varie de 5 à 60 %, tenant
en grande partie à la difficulté de définir la neuropathie diabétique.
Il existe un lien entre l'hyperglycémie et les
polyneuropathies qui apparaissent le plus souvent 5 à 10 ans après le début
du diabète et les traitements qui maintiennent une glycémie relativement
normale peuvent faire régresser des signes neurologiques, comme, par
exemple, les douleurs et sous insulinothérapie.
4.3.2. La polyneuropathie alcoolo-carentielle
Elle affecterait plus de 10 % des alcooliques
chroniques, Elle est cependant probablement en voie de diminution compte
tenu des modifications comportementales vis-à-vis de l’alcool. Elle doit
rester un diagnostic d’élimination.
Elle est habituellement secondaire à la toxicité directe de
l’alcool, beaucoup plus rarement à une carence en thiamine (vitamine B1)
qui donne des tableaux moteurs aigus. Elle touche les fibres motrices,
sensitives et végétatives.
Elle réalise une polyneuropathie distale
sensitivo-motrice et trophique des membres inférieurs (douleurs distales,
hypoesthésie superficielle en chaussette, déficit des muscles de la loge
antéro-externe entraînant un steppage à la marche, troubles trophiques
importants, rétraction tendineuse) par dégénérescence axonale.
4.3.3. Les carences en vitaminique
Les carences en vitamine B1, comme le béribéri par
carence d'apport et les déplétions par vomissements prolongés, donnent des
tableaux similaires :
La carence en vit.B6 (pyridoxine) donne une polynévrite
sensitive des patients traités par de fortes doses d'INH.
4.3.4. Les intoxications
Professionnelles
Elles peuvent être dues au plomb, à l'acrylamide (ciment
chimique, chez les ouvriers du bâtiment), au mercure, au sulfure de carbone
(vulcanisation du caoutchouc, soie artificielle), à l’arsenic, mais aussi
aux insecticides (DDT), aux solvants (trichloréthylène)
Médicamenteuses
Le plus souvent, elles sont dues aux produits suivants.
4
L'isoniazide (Rimifon™) par carence en vitamine B6
4
Nitrofurantoïne (Furadoine™) responsable d'une PN
sensitivo-motrice favorisée par l'insuffisance rénale.
4
Les alcaloïdes de la pervenche : vinblastine
(Velbé™) et surtout vincristine (Oncovin™) entraînant une neuropathie
axonale avec paresthésies et aréflexie et secondairement une atteinte
motrice.
4
Les taxanes : Taxol™ et Taxotère™
Plus rarement en cause les médicaments :
4
Autres anticancéreux : cisplatine
(Cysplatyl™), procarbazine (Natulan™)
4
Cardio-vasculaires : maléate de péhexilline
(Pexid™), amiodarone (Cordarone™)
4
Rhumatologie : sels d'or, chloroquine
4
Antibiotiques : métronidazole (Flagyl™),
chloramphénicol, éthambutol, streptomycine
4
Anti-épileptiques : barbituriques,
phenylhydantoïne (souvent limité à une aréflexie)
4
Respiratoires : almitrine (Vectarion™)
4.3.5. Les dysglobulinémies
4
La gammapathie monoclonale de signification
indéterminée « MGUS » IgG ou IgA.
Il se caractérise par des
mutations génétiques des plasmocytes. Les plus fréquemment rencontrées
sont :
Des réarrangements 14q32
(surtout t(11;14), plus rarement autres translocations -13/13q-
Une hyperméthylation
promoteur p16, p15
Une instabilité du
caryotypique.
La « MGUS » s’observe chez les personnes âgées et
serait relativement fréquente : 3% après 70 ans. Son incidence
augmente avec l’âge.
La gammapathie monoclonale est définie par la présence dans le sang
d’une protéine monoclonale « M » qui est produite par un clone
mineur de plasmocytes. Par définition, la MGUS présente les
caractéristiques suivantes.
Le taux sanguin de la protéine « M » est inférieur à
30 mg/l.
Il y a moins de 5 % de plasmocytes dans la moelle osseuse.
Il n’y a pas de complications. C’est l’absence de lésion osseuse,
d’insuffisance rénale, d’anémie ou d’hypercalcémie qui un des critères de
diagnostic.
Une faible quantité de protéine « M » dans les urines
(< 500 mg/24 heures).
4
Neuropathie démyélinisante à activité
« anti-MAGLa » avec présence d’une IgM mise en évidence par
immunoélectrophorèse doit être recherchée devant une neuropathie sensitive
ataxiante comportant une aréflexie et un tremblement d’attitude. L’EMG
objective une atteinte démyélinisante. Celles-ci sont volontiers associées
à une activité anticorps dirigée contre certains constituants de la
myéline, comme une activité anti-MAG.
Cas
de la patiente de 84 ans
4
Myélome soit dans sa forme lytique (atteinte
axonale à l’EMG), soit dans sa forme ostéocondensante (atteinte
démyélinisante à l’EMG)
4
Syndrome POEMS
Le syndrome « POEMS » ou maladie de Crow Fukase associe, dans sa forme
complète, cinq types d’anomalies dont les premières lettres donnent
l’acronyme de la maladie.
Polyneuropathie :
atteinte neurologique motrice et sensitive,
Organomégalie :
augmentation de taille des organes,
Endocrinopathie :
atteinte de plusieurs glandes endocrines comme la tyroïde, les îlots de
Langerhans (partie pancréas secrétant l’insuline) ou les gonades (ovaires
ou testicules),
M : dyscrasie
plasmocytaire (protéine M) qui correspond parfois à un myélome ou à
un plasmocytome solitaire
S (Skin) : présence de lésions ou d’anomalies cutanées.
4
Maladie de Waldenström
4
Cryoglobulinémies devant faire rechercher une
infection par le virus de l’hépatite C
4.3.6. Les maladies de système
4
Sjögren,
4
Mononeuropathies multiples par vascularite, comme
le lupus, la PAN et la sarcoïdose Lupus
4.3.7. Les autres pathologies malignes
4
Les hémopathies par infiltration leucosique et les
lymphomes (méningoradiculites par infiltration méningée), plexopathie
douloureuse asymétrique
4
Cancers par infiltration, plexopathie douloureuse
asymétrique ou atteinte paranéoplasique ; à rechercher devant une
neuropathie douloureuse (Denny-Brown) qui est une mono neuropathie
sensitive multiple.
4.3.8. Les autres étiologies
Les causes Infectieuses
4
Lèpre (voyage en pays d’endémie) qui est une mono
neuropathie sensitive multiple.
4
SIDA (polyneuropathies sensitives à la phase
tardive de l’infection).
L’amylose primitive ou secondaire (gammapathie)
Les neuropathies amyloïdes familiales :
Portugaise-type I, Ces affections autosomiques familiales débutent autour
de 30 ans. Elles réalisent une neuropathie sensitivo-motrice et végétative
évoluant vers la cachexie et la mort en une quinzaine d'années.
Les neuropathies héréditaires Dégénératives
Les neuropathies motrices et sensitives héréditaires
(NMSH)
4
De type I : forme hypertrophique de la Maladie
de Charcot Marie et Tooth. C’est la plus fréquente, débutant à
l'adolescence, de transmission le plus souvent autosomale dominante,
associée à une hypertrophie des troncs nerveux et un ralentissement des
vitesses de conduction nerveuse ;
4
De type II : forme axonale de la maladie de
Charcot-Marie-Tooth. Elle se différencie du type I par un début plus tardif
et l'absence de ralentissement des VCN.
4
De type III ou maladie de Déjerine-Sottas. C’est
une forme sévère du type I à début plus précoce et génétiquement
hétérogène.
Les neuropathies héréditaires sensitives et autonomes
(NHSA)
4
De type I ou maladie de Thévenard qui est une
affection autosomique héréditaire qui se manifeste vers la 3è décennie. Sa
traduction essentielle est la présence de maux perforants plantaires et une
hypoesthésie thermique et douloureuse en chaussette.
4
De type II ou maladie de Morvan qui est proche de
la précédente mais de transmission autosomale récessive et à révélation
plus précoce.
4
De type III (dysautonomie familiale ou syndrome de
Riley-Day).
5. Les
neuronopathies
5.1. Définition
Elle touche le ganglion rachidien situé dans la racine
postérieure. Elles sont donc purement sensitives.
5.2. Tableau clinique
Les signes cliniques sont
4
Des fourmillements des extrémités, des paresthésies
douloureuses asymétriques et distales
4
Une perte, variable, de toutes les sensibilités
avec souvent signes d’ataxie
4
Aréflexie diffuse
Son évolution peut être plus ou moins rapide.
Le diagnostic est
établi à partir de l’EMG.
5.3. Les étiologies
Les causes principales sont :
4
Un syndrome paranéoplasique associé à une petite
tumeur primitive dont la croissance est lente. Le cancer du poumon, en
particulier à petites cellules. La recherche d’anticorps IgG contre Hu
(anti-Hu). L’antigène Hu est une protéine du noyau du neurone de 35 à 40 kD
cancer le plus souvent en cause.
4
Le syndrome de Sjögren (biopsie des glandes
salivaires accessoires positives et négativité fréquente des auto-anticorps
SS-A et SS-B)
Cas du
patient de 62 ans
6. Polyradiculonévrites
6.1. L’approche étiologique
Tout d’abord, il faut se rappeler qu’elles quasiment
toujours idiopathiques. Elle se fait par rapport aux critères suivants.
4
Le contexte
o
antécédents familiaux de neuropathies périphériques
o
les risques professionnels (exposition à des
toxiques)
o
prises médicamenteuses neurotoxiques
o
maladies métaboliques et générales
4
Le type évolutif
o
évolution aiguë (plusieurs jours, quelques
semaines)
o
évolution subaiguë (plusieurs semaines, quelques
mois)
o
évolution chronique (plusieurs mois, quelques
années)
4
La topographie
o
atteinte bilatérale et symétrique
4
L’examen de l’EMG et de la stimulodétection
o
neuropathie axonale
o
neuropathie démyélinisante ou myélinique
4
La composition du LCS
o
hyperprotéinorachie avec dissociation
albuminocytologique, parfois avec hypercellularité
o
souvent normal
6.2. Le syndrome de Guillain Barré
6.2.1. Généralités
C’est une polyradiculonévrite inflammatoire aiguë avec
démyélinisation segmentaire multifocale d'origine auto-immune. On estime
l’incidence à 1/100 000. Il est rare chez le petit enfant.
Il existe fréquemment un antécédent infectieux
respiratoire ou digestif (55 %) dans les 15 jours précédents. Campylobacter jejuni, VIH et
Cytomégalovirus sont plus particulièrement impliqués.
Les lésions démyélinisantes seraient en rapport avec la
production et le passage dans les espaces endoneuraux d'anticorps dirigés
contre certains antigènes de la myéline, dont la nature n'a pas encore été
établie chez l'homme. L’infection virale déclenche une immunisation croisée
contre les antigènes du système nerveux périphérique.
6.2.2. La polyradiculonévrite aiguë évolue en trois phases
La phase
d'installation (1 à 4 semaines) souvent marquée par l'apparition de
paresthésies des extrémités associées parfois à des douleurs (rachialgies,
myalgies, radiculalgies). Secondairement apparaît le déficit moteur :
fatigabilité à la marche, paraparésie. L'atteinte motrice peut rester
cantonnée aux membres inférieurs ou évoluer de manière ascendante touchant
les membres supérieurs, le tronc et la face (la diplégie faciale étant
l'atteinte la plus évocatrice).
La phase en
plateau (une à plusieurs semaines) ou le déficit moteur est fixé
selon une intensité variable allant de la paraparésie simple à la
quadriplégie totale chez un patient intubé, ventilé et alimenté par sonde
gastrique en réanimation.
La phase de
récupération (un à trois mois) est observée dans environ 80% des cas,
mais il faut savoir :
ð
qu'une réanimation respiratoire est nécessaire dans
15% des cas
ð
que cette maladie est mortelle dans 5 à 10% des cas
ð
que des séquelles sont possibles : déficit moteur
(steppage)
ð
qu'une rechute peut survenir dans 5% des cas.
6.3. Les autres formes aiguës d’atteinte périphérique
6.3.1. La maladie de Lyme
Elle peut donner une méningoradiculite est secondaire à
une piqûre de tique. Elles sont atypiques, hyperalgiques, peu symétriques.
Et exceptionnellement motrices
L'interrogatoire recherche la notion d'érythème au point
de piqûre.
Une hypercytose est observée dans le LCS .La sérologie
de Borrelia burgdorferi
(spirochète) fait le diagnostic.
6.3.2. Les porphyries
Les porphyries sont des maladies génétiquement
déterminées, intéressant le métabolisme de l'hème. Les troubles
neurologiques s'observent essentiellement dans la porphyrie aigue
intermittente et variégata.
Le diagnostic repose sur :
4
Le déclenchement
o
La prise de certains médicaments : sédatifs,
anticonvulsivants, sulfamides, antibiotiques, œstroprogestatifs
o
Dans certaines situations : infection,
grossesse, excès d'alcool.
4
L'existence d'autres manifestations cliniques
o
Troubles psychiques : confusion, troubles du
comportement
o
Crises d'épilepsie,
o
Crises douloureuses abdominales.
4
L'EMG montre une atteinte axonale sans
ralentissement des vitesses de conduction.
4
La couleur brun rouge des urines lorsqu'elles sont
laissées à l'air pendant 24 H.
4
Les tests biologiques montrent une élévation dans
les urines des concentrations des précurseurs de l'hème (porphobilinogène,
uroporphyrines, acide delta-amino-lévulinique) et baisse de l'activité
uro-synthétase dans les globules rouges.
6.3.3. Le botulisme
Atteinte de la jonction neuromusculaire Due à une toxine
qui se développe électivement dans les conserves et provoque initialement
des troubles digestifs (paralysie du pharynx, bouche oesophagienne), des
signes neurovégétatifs (sécheresse des muqueuses), une paralysie de
l'accommodation (mydriase aréflexique), puis une polyneuropathie motrice.
6.4. La diphtérie (bacille de Löeffler)
Un à trois mois après l'angine, apparition d'une
paralysie du voile du palais, puis de l'accommodation puis une neuropathie
démyélinisante sensitivo-motrice des membres inférieurs régressive sur 3
mois.
7. Les
mononeuropathies
7.1. Le tableau clinique
Il est dominé par l’asymétrie des lésions C’est un
syndrome neurogène asymétrique. Plusieurs types sont à envisager :
4
Une atteinte unique è tronculaire
4
Une atteinte multiple è
tronculaire è
mononeuropathies multiples
7.2. Les vascularites
7.2.1. Les artérites nécrosantes
Elles sont liées à la formation de complexes immuns
circulants. Ils peuvent survenir de novo, comme dans la périartérite
noueuse ou être secondaires à une maladie générale.
Elles sont caractérisées par une nécrose de la paroi
vasculaire avec infiltration leucocytaire, hémorragie, dépôts fibrinoïdes. Elles entraînent une neuropathie par un
mécanisme ischémique (véritable accident vasculaire du nerf).
Le contexte est celui d'une maladie systémique (2/3 des
cas) avec fièvre, perte de poids, asthénie, manifestations articulaires,
cutanées et rénales, syndrome douloureux...
Dans 2 cas sur 3, le tableau neurologique est celui
d'une mononévrite multiple
d'installation brutale ou d’une multinévrite. Une atteinte dans les
territoires suivants est plus fréquente :
4
du sciatique poplité externe (75%),
4
du cubital (25%),
4
du sciatique poplité interne, du médian, du crural
et du radial (10% pour chacun de ces territoires).
4
Les examens neurophysiologiques orientent vers une
neuropathie axonale et l'étude du LCS est normale.
Cas de la
patiente de 26 ans
7.2.2. Les vascularites lymphocytaires
Elles peuvent accompagner certaines maladies
parasitaires ou infectieuses comme les infections à rétrovirus. Leur rôle
dans le développement de la neuropathie est moins clairement établi.
8. Conclusions :
points essentiels à retenir
Neuropathies Périphériques :
Signes cliniques : atteinte motrice, sensitive
et/ou végétative, électriques, biologiques et histologiques résultant d'une
atteinte du neurone périphérique.
Importance de l’EMG : Les vitesses de conduction
« VC » reflètent le processus en cause
4
Neuropathies démyélinisantes è
ralentissement des VC motrices et sensitives
4
Neuropathies axonales è
normalité des VC, baisse d'amplitude des réponses motrices et sensitives.
4
Subaiguë et chronique et longueur-dépendante è
polyneuropathie toxique (médicaments, alcool), métabolique (diabète,
atteinte rénale) principalement
4
Chronique è neuropathie héréditaire
Les polyradiculonévrites è
tableau symétrique
4
Aiguë et démyélinisante è
polyradiculonévrite (Syndrome de Guillain Barré)
Les mononeuropathies è
tableau asymétrique
4
Une atteinte unique : tronculaire
4
Une atteinte multiple : tronculaire è
mononeuropathies multiples
o
Maladies générales
o
Diabète
Syndromes canalaires (non traités durant la séance)
Compressions des nerfs périphériques (lieux de passage
compressions dites posturales. Démyélinisation segmentaire (EMG)
4
Nerf médian au canal carpien :
o
Paresthésies ou engourdissement nocturne, des trois
premiers doigts
o
Signes de Tinel et de Phalen
o
Traitement : infiltration de corticoïdes dans
le canal carpien, chirurgie si échec
4
Nerf ulnaire dans la gouttière
épithrochléo-oléocrânien
o
Paresthésies des 4ème et 5ème doigts, hypoesthésie
territoire du nerf à la main
o
Déficit moteur des muscles intrinsèques de la main
o
Traitement chirurgical (transposition du nerf)
4
Nerf radial dans la gouttière humérale (fractures
de l’humérus et compression externe du bras)
4
Nerf fémoro-cutané ou méralgie paresthésique
4
Nerf fibulaire au col de la fibule
o
Déficit des muscles de loge antéro-externe jambe
(steppage)
o
Déficit sensitif discret du dos du pied
o
Traitement conservateur

|