|
EPU95 – Montmorency
Neurologie
 Mise à jour du 24 Avril 2007 Mise à jour du 24 Avril 2007 

Les Maladies à
prions
Pr.
D. Dormont
Chef du service de
neurovirologie - Centre de recherche du CEA à Fontenay aux Roses
Séance du 7 février 2002
1.
Introduction
Les
Encéphalopathies Subaiguës Spongiformes Transmissibles (ESST), que l’on
appelle de manière impropre maladies à prions, puisque le prion est une
notion purement théorique qui n’a pas encore été formellement démontrée,
sont des maladies rares. Ces maladies à prions touchent l’homme ou
l’animal.
1.1. Chez
l’homme
Quatre
maladies appartiennent à ce groupe :
4 Le Kuru,
4 La maladie de
Creutzfeldt-Jakob (MCJ),
4 Le syndrome de
Gerstmaen-Straüssler-Spheinker
4 L’insomnie fatale.
1.2. Chez
l’animal
Il
y a cinq maladies qui appartiennent au groupe des ESST.
1.2.1. La
tremblante naturelle du mouton et de la chèvre
1.2.1.1. La
maladie
C’est
une vieille maladie connue depuis 1732. Elle est présente partout dans le
monde sauf dans deux pays qui ont réussi à l’éradiquer, l’Australie et la
Nouvelle-Zélande.
Cette
maladie peut être présente avec une incidence très élevée : certains
troupeaux du bassin du Sud-ouest de la France ont parfois jusqu’à 15 à 20 %
des animaux qui développent une tremblante.
1.2.1.2. 1ère
démonstration de transmissibilité
Deux
vétérinaires français, Cuiller et Chelles, en 1936, ont prélevé le cerveau
d’un mouton malade. Ils en ont fait un broyat qu’ils ont filtré, puis ils
ont inoculé directement dans le cerveau de moutons sains un aliquot de ce
broyat. Les animaux inoculés ont développé, 2 ans plus tard, une maladie en
tout point identique à la maladie naturelle.
1.2.2.
L’encéphalopathie spongiforme bovine
C’est
une maladie apparue au début des années 80 au Royaume-Uni.
1.2.3.
L’encéphalopathie féline spongiforme
C’est
une maladie qui concerne les chats (99 cas répertoriés dans le monde, 97
sont sur le territoire du Royaume-Uni, 1 au Lichtenstein, 1 en Norvège).
Cette
maladie féline est liée à la contamination des chats par l’agent de
l’encéphalopathie bovine spongiforme, contamination qui s’est effectuée
probablement au travers de l’alimentation ; ceci démontre que les
agents des ESST peuvent être pathogènes dans des espèces qui ne sont pas
leur espèce d’origine, et donc capable de franchir les barrières d’espèces.
1.2.4.
L’encéphalopathie transmissible du vison
C’est
également un exemple de franchissement de barrière d’espèce : il existe
des fermes qui élèvent des visons pour leur fourrure. Les visons sont des
carnivores, et les éleveurs leur donnent à manger les carcasses non
exploitables d’ovins ou de bovins dans les abattoirs. Si la carcasse de
vache ou le mouton provient de bête infectée par l’agent de la tremblante
ou par l’agent de l’encéphalopathie bovine, le vison en 7 mois en moyenne
développe une encéphalopathie dont il va mourir. Comme les visons sont à
plusieurs dans le parcage et qu’ils sont carnivores, lorsqu’un animal meurt
dans une cage, il est mangé par les autres animaux de la cage qui à leur
tour se contaminent.
Ceci
explique la disparition d’élevage entier de visons en Finlande, en
Allemagne, en ex-URSS, au Canada, et aux USA.
1.2.5. Le
dépérissement chronique des ruminants sauvages
Cette
maladie étonnante est la seule maladie à prion chez les animaux sauvages.
Elle touche l’élan, le cerf, le daim, les grands ruminants sauvages dans
les forêts de deux Etats américains, le Wyoming et le Colorado. Cette
maladie est en train de monter dans le Nord des USA et le Sud du Canada où
quelques cas viennent d’être diagnostiqués. Elle pose trois
problèmes :
4 Premier problème :
Comment un prion se transmet en dehors de tout contexte domestique et de
toute intervention humaine ?
4 Deuxième question :
Sur quel élément connaît-on que l’incidence augmente ? Dans ces Etats,
un chasseur qui tue un cerf ou un gros ruminant sauvage a l’obligation
d’apporter la tête aux autorités qui pratiquent un examen
neuro-pathologique. Avec ce système de surveillance extrêmement fruste,
passif et peu performant, les incidences de la maladie ainsi mesurées se
situent entre 8 et 15 %. Même en tenant compte des biais intrinsèques de ce
type d’évaluation, la prévalence de la maladie est quelque chose d’important
dans cette population.
4 Troisième
question : Est-ce dangereux pour l’homme ? Comme pour les autres
maladies du groupe touchant les grands ruminants.
2.
Caractéristiques des
ESST
2.1. Généralités
Que
ces maladies soient naturelles ou expérimentales, qu’elles soient humaines
ou animales, elles ont en commun un certain nombre de caractéristiques
communes :
4 Elles sont
transmissibles à des animaux de laboratoire.
4 Quand le sujet est
infecté, une période d’incubation asymptomatique, extrêmement longue au
regard de l’espérance de vie. Et chez l’homme, la période d’incubation la
plus longue connue est de 47 ans.
4 Quand les signes
cliniques apparaissent, ils sont la traduction unique de la dégénérescence
du système nerveux central (SNC). En d’autres termes, il n’y a pas
d’atteinte vasculaire, ni cardiaque ou hépatique, ni d’atteinte des nerfs
périphériques sauf dans de rares cas japonais.
4 L’évolution se fait sur
un mode subaigu sans rémission en 6 semaines à 6 mois en moyenne chez
l’homme.
4 A l’autopsie, on
constate que les lésions sont limitées au SNC et faites d’une triade
caractéristique :
o
Mort
neuronale massive
o
Vacuolisation
des neurones
o
Prolifération
des astrocytes et activation des astrocytes, appelée astro-gliose ou
hyperastrocytose d’activation.
2.2. L’anatomie pathologique
du SNC
2.2.1. Les
vacuoles disséminées
Elles
correspondent à une « gliose ». Elles sont mises en évidence par
un immuno-marquage du protéino-glio-fibrillaire acide (GFAP). Celui-ci est
un marqueur d’activation des astrocytes.
2.2.2. Plaque
de dépôts amyloïdes
Ce
sont ces plaques que l’on retrouve dans le vieillissement spontané, dans la
maladie d’Alzheimer, mais dont la morphologie fine et la composition
chimique est ici très différente.
4 Il n’existe aucun des
signes que l’on voit habituellement dans les maladies infectieuses :
o
Pas
de syndrome inflammatoire biologique,
o
Pas
de fièvre,
o
Absence
de réponse du système immunitaire,
o
Absence
d’atteinte des gaines de myéline dans le SNC.
4 Ces ESST sont des
maladies lentes sans démyélinisation et sans œdème cérébral, qui sont
pourtant les signes caractéristiques des infections virales lentes du SNC.
4 Elles ne s’accompagnent pas d’afflux dans le SNC
de cellules immunocompétentes. Le système immunitaire ne répond pas à
la présence de l’agent, il n’y a pas de synthèse d’anticorps, donc pas de
test de dépistage simple, et pas de réponse immune cellulaire qui puisse
être mis en évidence chez les sujets qui sont infectés. Ceci est un
paradoxe, car le système immunitaire est une cible de la multiplication des
prions.
Comment
expliquer que le système immunitaire ne réponde pas, alors même qu’il est
infecté ?
Jamais
personne n’a vu un prion, quel que soit le microscope utilisé.
Jamais
on n’a vu un élément qui puisse ressembler de près ou de loin à un
parasite, un champignon, un virus ou une bactérie, ceci parfois malgré un
titre infectieux faramineux. Par exemple, lorsque le hamster
expérimentalement inoculé par la tremblante du mouton, il a 1011 unités
infectieuses par gramme de tissu cérébral, c’est-à-dire qu’avec 1 gramme de
tissu cérébral on pourrait tuer 100 milliards de hamsters sains.
Ce
sont des maladies qui peuvent incuber plusieurs dizaines d’années chez
l’homme, pour lesquelles on n’a pas de test de diagnostic, pas de
thérapeutique, et pour lesquelles on ne connaît pas l’agent étiologique,
même si des hypothèses intéressantes et novatrices peuvent être proposées.
D’une
façon générale et sans préjuger de l’avenir, dans l’état actuel des
connaissances, ces maladies sont, tout au moins pour les maladies humaines,
des maladies transmissibles mais pas contagieuses.
La
seule anomalie biologique que l’on peut mettre en évidence dans ces
maladies est une anomalie du métabolisme de certaines protéines. Le sujet
infecté va accumuler, certaines de ces protéines et parmi ces
protéines :
1. Trois s’accumulent,
indépendamment du titre infectieux, mais qui sont intéressantes sur le plan
physiopathologique :
o
L’apolipoprotéine
E, ayant une importance dans certaines formes génétiques de maladie
d’Alzheimer.
o
La
cathepsine D
o
La
glutamine synthétase qui est une enzyme importante dans la compensation du
stress oxydatif. On sait qu’il n’y a pas de mort neuronale sans stress
oxydatif. Il y a donc une certaine logique à ce que cette glutamine
synthétase soit hypersollicitée.
2. Une protéine ne
s’accumule que dans les ESST et proportionnellement au titre infectieux.
C’est la Protéine P ou PrP-res pour protéine P résistante aux protéases.
Dans l’état actuel des connaissances :
o
Cette
protéine s’accumule proportionnellement au titre infectieux.
o
Elle
se « co-purifie » avec l’agent infectieux, c’est-à-dire qu’à
chaque fois que l’on essaye de purifier l’agent infectieux, on purifie
cette protéine. Elle est le constituant majeur des fractions infectieuses.
Quand on enlève la protéine des fractions infectieuses, l’infectiosité
disparaît.
o
Tout
se passe comme si la protéine était soit l’agent infectieux, soit
intimement associé à l’agent infectieux.
La seule anomalie
détectable et spécifique des ESST est donc l’accumulation d’une forme
anormale d’une protéine de l’hôte, la PrP.
3.
ÉVOLUTION
des Connaissances sur la PROTÉINE PrP
3.1. La Protéine PrP
du sujet sain, dite PrP-c
Tous
les mammifères ont une PrP. L’homme en a donc une. On ne connaît pas de PrP
en dehors des mammifères. La protéine normale de l’homme, sans rentrer dans
les détails de la formule, présente :
4 Des répétitions
d’octapeptides qui fixent le cuivre,
4 Un domaine hydrophobe,
4 Un pont disulfure,
4 Deux sites de
glycosylation , ce qui permet de détecter 3 formes de protéine suivant
qu’elle est non-glycosylée, ou mono-glycosylée ou bi-glycosylée.
4 A sa portion
C-terminale, une sérine qui va permettre de fixer une structure
glucido-lipidique, appelé GPI (glycosylphosphatidyl inositol) ; grâce
à cette structure la protéine va s’accrocher à la face externe de la
membrane cellulaire.
C’est
une protéine qui est à l’extérieur
de la cellule, accrochée à la membrane cellulaire. Cela lui permet, en
théorie, d’être soit un récepteur, soit une molécule d’interaction avec la
matrice extra cellulaire.
3.2. La Protéine PrP
du sujet infecté, dite PrP-res
Quand
la protéine PrP a été découverte par S. Prusiner, au début des années 1980
(Prix Nobel en 1997), la première question posée fut : qu’est-ce qui
diffère entre la protéine normale présente chez tous les hommes et la
protéine qui va s’accumuler chez les patients infectés ?
La 1ère hypothèse
soulevée fut : ne s’agit-il pas d’une modification de la séquence
primaire de la protéine (modification de la composition chimique
élémentaire de la protéine) ?
Cette
hypothèse est mauvaise : la protéine pathologique a exactement la même
composition chimique en acides aminés et aux mêmes positions que la
protéine normale.
Comment expliquer qu’une
protéine, qui ne modifie pas sa structure élémentaire, se mette à
s’accumuler ?
Il
faut postuler qu’elle acquiert de nouvelles propriétés physico-chimiques.
L’immuno-détection
par western-blot de la PrP normale à l’aide d’un anticorps spécifique est
facilement réalisable à partir d’un homogénat de cerveau sain. Sur
l’électrophorèse apparaissent à des niveaux différents, la forme
non-glycosylée, la forme mono-glycosylée et la forme bi-glycosylée de la
protéine.
4 Si cet examen est
pratiqué sur un homogénat de cerveau sain qui a été traité auparavant par
une enzyme qui dégrade les protéines (la protéinase K), à l’électrophorèse
il n’y a plus de signal correspondant à la protéine.
4 La protéine normale est
digérée par les enzymes protéolytiques. En revanche la protéine
pathologique, non seulement s’est accumulée (augmentation du signal), mais
de plus elle est devenue résistante aux protéases.
Comment expliquer cette
résistance aux protéases ?
L’hypothèse
la plus probable, c’est que la PrP a simplement modifié sa forme dans
l’espace. Si cette hypothèse est vraie (c’est l’hypothèse du prion et il y
a beaucoup d’arguments qui la supportent), la protéine n’est absolument pas
modifiée dans sa composition chimique élémentaire, elle change de forme
dans l’espace. Ce simple changement de conformation suffit à en faire un
agent infectieux. On rentre dans un nouveau domaine de la médecine et de la
biologie, et si cette hypothèse se confirme, cela changera beaucoup de nos
concepts.
Deux
mécanismes sont possibles pour qu’une protéine s’accumule :
4 Soit le gène
s’hyper-exprime
4 Soit il y a une anomalie
du catabolisme.
Le
gène de la Protéine PrP est connu (appelé PRPN). Il est situé sur le bras
court du chromosome 20. C’est un gène très rustique : 2 exons séparés
par un intron (10 kilobases) et toute la séquence codante est dans le 2ème
exon.
-
Ce
gène est exprimé majoritairement dans le SNC (50 fois plus dans les
neurones que dans les cellules gliales)
-
En
dehors du SNC, il est exprimé dans le système immunitaire surtout mais à
des niveaux plus réduits.
Dans
les modèles animaux, le gène n’intervient pas dans le mécanisme qui conduit
à l’accumulation de la protéine.
-
Quand
on quantifie chez les animaux infectés l’accumulation de la protéine
pathologique, PrP-res (résistante aux protéases) ou PrP-sc (scrapie pour
tremblante en anglais) et que l’on mesure l’expression du gène, on se rend
compte que la protéine s’accumule sans modification de l’expression du
gène.
-
il
s’agit donc d’une anomalie post transcriptionnelle, au delà de la mécanique
d’expression des ARN et de leur lecture par les ribosomes.
4. la Maladie Creutzfeld
-Jakob
Chez
l’homme, en particulier dans la maladie de Creutzfeld Jakob, il existe 3
formes étiologiques
4 La Forme sporadique : c’est-à-dire sans lien
entre les patients, sans lien entre le patient et l’environnement :
apparaissant chez l’homme et la femme à égalité entre 60 et 70 ans. Plus de
90 % des MCJ sont sporadiques.
4 La Forme familiale : elle représente 5 à 10
% des MCJ. On note l‘existence de familles dans lesquelles à chaque
génération 50 % des sujets sont atteints. Trois foyers ont été décrits (2
en Tchécoslovaquie, et 1 en Israël chez des juifs d’origine libyenne). Son
apparition est plus précoce dans la vie (45 à 55 ans)
4 La Forme
iatrogénique : elle est acquise au cours d’un acte médical ou
chirurgical.
Ces
trois formes sont transmissibles à l’animal, même la forme génétique. C’est
le seul exemple en médecine d’une maladie génétique transmissible
horizontalement.
4.1. Forme Sporadique
de MCJ
Dans
la forme sporadique, la PrP a une séquence normale, et un jour pour une
raison que l’on ne connaît pas elle va changer de forme, s’accumuler et
provoquer la démence.
4.1.2.
Epidémiologie
C’est
une maladie rare. Sur 10 000 français qui décèdent 1 meurt de MCJ.
L’incidence est de 1 à 1,5 nouveaux cas par an et par million d’habitants
4.1.3.
Clinique
C’est
le tableau d’une démence pour laquelle il n’y a pas d’étiologie. La démence
est toujours au premier plan de la clinique à la phase d’état de la
maladie.
Deux
signes sont très fréquents :
4 L’ataxie cérébelleuse
qui existe pratiquement toujours
4 Les myoclonies qui
permettent le diagnostic, et que l’on ne sait pas traiter.
o
Au
début, elles sont déclenchées par l’examen médical : pincement d’un
doigt va donner des clonies du bras,
o
Ensuite
le même pincement entraînera des myoclonies de l’hémicorps
o
Ultérieurement,
le seul fait de rentrer dans la chambre déclenchera de grandes crises
généralisées de myoclonies.
Des
myoclonies sans fièvre, avec IRM normale chez un sujet de 65 ans doivent
faire suspecter une MCJ, surtout s’il y a une dégradation cérébrale.
4.1.4. Examens
complémentaires
EEG
Il
objective dans 60 % des cas, sur un fond de ralentissement général de
l’EEG, des complexes triphasiques lents de 1,5 hertz de périodicité que
l’on appelle des anomalies pseudo périodiques. Elles sont diffuses à
l’ensemble des dérivations de l’EEG. Cet aspect chez un sujet qui commence
à devenir dément en dehors de toute intoxication au CO, est un argument
fort pour une MCJ.
L’étude du LCR
Elle
permet de mettre en évidence à la période symptomatique la protéine 14-3-3.
C’est une protéine associée à la mort neuronale quand celle-ci est
importante :
4 On peut la détecter dans
des maladies autres que la MCJ, par exemple au décours d’un accident
vasculaire cérébral, dans certaines formes d’épilepsie, dans certaines
formes de maladies d’Alzheimer, dans les encéphalopathies herpétiques.
4 Quand la clinique est
évocatrice d’une MCJ, la présence d’une protéine 14-3-3 positive permet
pratiquement d’affirmer le diagnostic.
4.2. Forme Familiale
de MCJ
Elles
sont toutes liées à une mutation dans le gène qui code la protéine PrP, et
qui est appelé PRPN chez l’homme.
L’individu
naît avec une protéine mutée. Cette protéine va fonctionner correctement
pendant quelques dizaines d’années et puis elle va changer de forme,
s’accumuler et provoquer la démence.
4 Les mutations retrouvées
sont multiples. Leur pénétrance est de 1, c’est-à-dire que dès lors qu’un
sujet est porteur de la mutation, il développera tôt ou tard la MCJ.
4 La mutation au codon 178
entraîne une forme familiale de MCJ , se traduisant par une démence.
Il
est intéressant de rapprocher l’Insomnie Fatale de la forme familiale de
MCJ.
4.3. L’insomnie Fatale
L’insomnie
fatale est « fascinante », car elle est rare, 7 familles connus
dans le monde.
C’est
une insomnie rebelle à toute thérapeutique, confirmée à l’EEG des 24 heures
et cet EEG objective une disparition sélective du sommeil paradoxal pendant
quelques semaines au moins.
Elle
évolue rapidement vers des troubles de l’équilibre, des troubles de
l’articulation des mots, puis un état hallucinatoire extrêmement riche au
plan onirique, puis le coma et la mort en 13 mois en moyenne.
C’est
une insomnie mortelle dans 100 % des cas. Elle est liée à une mutation du
gêne de la PrP et il s’agit de la même mutation que celle de la MCJ
familiale au codon 178. Même substitution de base sur l’ADN, donc même
substitution d’acides aminés et pourtant dans un cas c’est une maladie
Creutzfeldt-Jakob, alors que dans l’autre cas c’est une Insomnie fatale.
C’est
une maladie génétique, mais elle est infectieuse puisqu’elle peut être
transmise à l’animal par inoculation d’un homogénat de cerveau du patient
décédé.
On
se trouve ici devant une même mutation du même gêne responsable de deux
tableaux cliniques différents.
Il
n’y a pas de traitement de la maladie à prions. Il existe quelques
molécules qui ont une petite efficacité in vivo dans les modèles d’animaux
sans en connaître le mode d’action :
4 la iodorubicine, les
sulfates de sextran
4 les dérivés de
l’amphotéricine B
Malheureusement,
il faut pour être efficace que le traitement débute dès que l’animal est
infecté, donc c’est inapplicable à l’homme, puisque le diagnostic de
l’affection n’est fait que tardivement, le jour où le sujet devient
symptomatique.
5. Le prion
5.1. Transmissibilité
Les
maladies à prions sont transmissibles, facilement au sein de la même
espèce, mais aussi transmissibles entre espèces différentes.
4 La MCJ humaine est
transmissible au singe, à la chèvre, au cochon d’inde, à l’hamster, à la
souris, au rat, au chat, au furet et au vison ce qui représente un vaste
spectre de transmissibilité interspécifique.
4 La transmissibilité
entre espèces différentes est plus difficile qu’au sein de la même espèce.
Il existe une barrière d’espèces que les agents sont capables de franchir.
4 Le déterminisme
moléculaire de la barrière d’espèce dépend de l’homologie de séquence
du gène de la PrP du donneur et du receveur. Plus ces gènes sont proches,
plus la transmission spécifique est facile ; plus ces gènes sont
différents et plus la transmission sera difficile.
5.2. La protéine P a
deux rôles
Le rôle
Normale, elle est le facteur de susceptibilité à
l’infection. Pathologique,
elle est certainement l’agent
infectieux lui-même.
Exemples :
-
Le
singe a une PrP qui est à 98 % homologue à celle de l’homme, la
transmission au singe réussit dans 95 % des cas.
-
La
souris qui a une PrP a 70 – 80 % identique à celle de l’homme, la
transmission homme-souris ne réussit que dans 15 % des cas.
Si
maintenant par les procédés modernes de la biologie, on introduit le gène
de la PrP humaine chez une souris (souris transgénique exprimant la PrP
humaine), l’animal transgénique devient sensible à 100% des cas à la MCJ.
Cela montre bien que le gène de la
PrP est bien le facteur nécessaire conditionnant la barrière d’espèce.
En
restant dans l’exemple homme-singe, si on prélève du cerveau d’un patient
atteint de MCJ et que l’on purifie les fractions infectieuses de la PrP,
c’est-à-dire la PrP pathologique humaine et si on l’inocule au singe. Le
singe, quelques années plus tard, développe une MCJ expérimentale.
Comme
dans toutes les maladies à prion, le singe
a accumulé la PrP pathologique, mais
la protéine accumulée n’est pas la protéine humaine qui a servi à
l’infecter. Le singe accumule sa propre protéine sous une forme
anormale. Il faut donc imaginer :
4 Un premier mécanisme,
par lequel la protéine humaine, qui sert d’agent infectieux pour le singe,
interagit avec son homologue simien et lui transfère son anomalie
4 Un 2ème mécanisme par
lequel cette première molécule de singe, rendue anormale, propage son
anomalie au sein de l’organisme infecté.
Et
nous pourrons dire, en toute rigueur, que la composition chimique
élémentaire de l’agent infectieux qui se développe chez le singe n’est pas
la composition chimique de l’agent infectieux qui a servi à le rendre
malade.
Pendant la
période asymptomatique, où se trouve l’agent infectieux ?
Est-ce
qu’on est dans une situation où l’agent est dans un réservoir, sans se
multiplier comme on le pense parfois dans certaines formes de
tuberculose ;
Est-ce
une situation semblable au VIH où l’agent se multiplie constamment en
créant des désordres que l’organisme au début compense ? C’est ce qui
se passe avec le prion.
Une
souris inoculée par voie intra péritonéale, par l’agent de la tremblante du
mouton. Dès le 4ème jour après l’infection, l’agent est dans le système
immunitaire, dans les organes lymphoïdes secondaires, et il se met à se
multiplier, jusqu’à la mort de l’animal. Ce n’est qu’à partir de la 2ème
moitié de la période d’incubation que l’agent commence à être détectable
dans le SNC. Une fois qu’il est dans le cerveau (neuro-invasion), la
réplication est quasi-exponentielle, mais les signes cliniques
n’apparaissent que 180 jours après l’infection. Avant que les signes
cliniques n’apparaissent, l’agent est présent à un haut titre dans le SNC
et en dehors du SNC au moins dans certaines maladies à prions.
5.3. Le degré
d’infectiosité des organes d’un sujet infecté
Le
classement de l’O.M.S. est fonction des organes atteints et des risques
infectieux potentiels. Elle prend en compte toutes les maladies à prion
connues, toutes les distributions d’infectiosité connues, le tout dans un
« un scénario catastrophe »
Ce
tableau récapitule les organes en fonction de leur catégorie pour
déterminer le risque potentiel d’infectiosité.
|
Classification OMS
|
|
4 Catégorie 1 :
haute infectiosité : atteinte du SNC (sans oublier la rétine et
l’oreille interne)
4 Catégorie 2 :
infectiosité moyenne : c’est le tissu lymphoïde secondaire (tube
digestif, rate)
4 Catégorie 3 :
infectiosité faible ou intermittente : les gros troncs nerveux, le
poumon, le foie, le pancréas.
4
Catégorie
4 : pas d’infectiosité détectée : le sérum, le muscle
squelettique, le lait è cela ne veut pas dire
qu’il n’y a pas de prion, mais insuffisamment pour établir une infection
chronique et une maladie.
|
5.4. Les paramètres de
la transmission
Les
paramètres sont nombreux et dépendent de :
- L’inoculum : la dose d’agent, la
souche d’agent, l’espèce donneuse,
- la voie d’inoculation : intra
cérébrale, injection ou instrumentale, voie orale
- la génétique de l’hôte et
particulièrement le gène de la PrP,
- l’état du système immunitaire.
|
Efficacité différentielle de
plusieurs voies d’inoculation
|
|
4
La voie intra cérébrale qui est de loin la plus
efficace
4
La voie intraveineuse est 7 à 10 fois moins
efficace que la voie intra cérébrale,
4
La voie sous cutanée est 25 000 fois moins
efficace que la voie intra cérébrale,
4
La voie orale est 125 000 fois moins efficace que
la voie intra cérébrale.
|
Ce tableau est valide, pour la maladie Creutzfeld-Jakob
classique. En revanche, il n’est certainement pas vrai pour la nouvelle
variante de MCJ, liée à l’agent bovin qui se propage beaucoup plus
facilement par voie orale.
Donc
à l’hôpital et en pratique médicale, il faut faire attention surtout dans
les actes qui donnent accès au système nerveux :
4 En neurochirurgie
(intervention stéréotaxique, greffe de dure mère,…)
4
En ophtalmo,
4
En ORL
4 En neurologie quand on
fait des ponctions lombaires.
5.5. La résistance de
l’agent infectieux
5.5.1. Le contexte
Un
des problèmes que pose ces maladies rares est la résistance des agents
infectieux aux méthodes qui habituellement inactivent les micro-organismes.
Aucune
des techniques, utilisant la chaleur sèche (le four, ne décontaminent
totalement un objet ou une matière infecté par ce type d’agent
infectieux :
-
à
180 ° pendant 24 h,
-
à
320 ° pendant 1h,
-
à
600° pendant 15 minutes.
5.5.2. Les
procédés efficaces
Il
existe trois procédures actines vis à vis de ces agents
infectieux :
-
L’autoclavage
(chaleur humide) pendant 18 minutes à 134° – 136°
-
La
soude normale pendant une heure à 20° C
-
L’eau
de Javel pure ou au demi, pendant 1 heure à 20° C.
Ce
sont des procédés extrêmement drastiques. Il ne faut pas se contenter du
« Poupinel » !
Deux
professions doivent prendre cela en compte :
-
les
neuro-chirurgiens, ophtalmo et ORL
-
les
dentistes qui doivent s’équiper en autoclave,
Si,
par hasard, cette n.v-MCJ commençait à se disséminer, les médecins
praticiens d’une façon générale qui utilisent des instruments devront
appliquer les procédures d’inactivation efficaces.
5.5.3. Les
formes de résistance
Comme
d’habitude, les micro-organismes arrivent à échapper à l’inactivation dans
certaines situations.
Par
exemple, le VIH, virus fragile à la chaleur, est en général détruit dès 56
°. Mais si le VIH, au lieu d’être mis dans un milieu de culture, est placé dans une solution très
concentrée en protéines, il faudra 45 minutes pour l’inactiver. Car
l’environnement protéique protège le virus d’une inactivation par la
chaleur. Pour les prions, c’est un peu semblable dans une autre
situation :
4 Quand on prend du prion
d’une MCJ, qu’on l’inactive suivant les recommandations de l’OMS (135-136°
pendant 18’ à l’autoclave), et qu’on inocule ensuite les animaux avec le
matériel traité, aucun animal ne tombe malade.
4 Si, avant la mise en
autoclave, on utilise la méthode qui se pratique dans tous les services
d’ana-path de France, c’est-à-dire d’immerger le cerveau dans du formol
pour le fixer et pour pouvoir l’examiner au microscope, à ce moment là
l’autoclave n’est plus efficace. Le pré-traitement par le formol inhibe les
effets ultérieurs de l’autoclave.
5.6. Les mécanismes
d’action du prion
Quand
on ne connaît pas l’agent infectieux, l’une des façons d’approcher sa
nature, c’est de lui appliquer des procédés d’inactivation physico-chimique
dont on connaît les mécanismes.
-
Chaque
fois que l’on applique des procédures qui ne touchent pas aux acides
nucléiques, mais qui découpent ou dénaturent les protéines en modifiant
leur forme, on perd l’infectiosité.
-
Quand
on applique des procédures qui altèrent les acides nucléiques sans toucher
à la structure tridimensionnelle des protéines, il n’y a pas d’effet sur
l’infectiosité.
Tout
se passe comme si les acides nucléiques n’avaient aucun rôle sur l’infectiosité.
Tout se passe comme si la structure tridimensionnelle des protéines était
le support de l’infectiosité.
5.6.1. Le rôle
de la Protéine PrP
Chez le sujet
normal
Le
rôle de cette protéine est mal connu. On a quelques éléments qui permettent
de penser qu’elle intervient :
-
dans
la transmission synaptique,
-
dans
la compensation du stress oxydatif,
-
qu’elle
interagit avec la matrice extracellulaire.
On
n’a pas une vraie vision de son rôle général dans le SNC.
Chez le sujet
infecté
La
protéine a trois rôles :
-
Elle
règle la barrière d’espèce.
-
Elle
règle la susceptibilité individuelle à l’infection par les prions.
-
Elle
a un rôle majeur dans la pathogénèse :
o
C’est
à cause de son changement de forme, que la protéine s’accumule.
o
C’est
à cause de cette accumulation dans la cellule neuronale que cette cellule
va mourir. Comme les neurones ne se régénèrent pas et que l’on n’est pas
capable de les régénérer, c’est à cause de cette accumulation de la
protéine que l’individu meurt.
Les
chercheurs suisses ont été les premiers à réaliser des souris « knock
out » pour le gène de la PrP, c’est-à-dire des souris génétiquement
manipulées qui n’expriment pas la protéine PrP. Ces souris n’ont aucune
protéine PrP :
-
Elles
ont un développement normal
-
Elles
apprennent un peu moins vite, mais la rétention du conditionnement est
correcte
-
Elles
n’ont pas de déficit neurologique
-
Mais
surtout elles ne sont pas infectables par les prions.
On peut en déduire que
pour être infectable par un prion, il faut extérioriser la protéine normale
à la surface de ses cellules. Ces souris ont été très
utiles pour appréhender certaines notions en santé publique que l’on
subodorait mais que l’on n’avait pas démontrées.
-
Des
souris « knock-out », auxquelles on inocule dans le cerveau un
prion (peu importe lequel), vont mourir de vieillesse (700 jours). Si, à
leur mort, le cerveau est recueilli pour être inoculé à une souris non
résistante au prion, cette dernière va être atteinte de la maladie.
-
Cela
veut dire que le prion est capable de rester infectieux 700 jours dans un
milieu biologique qui ne lui permet pas de se répliquer.
Métabolisme de
la Protéine PrP normale
La
protéine PrP est synthétisée dans le réticulum endoplasmique. Elle est
terminée dans l’appareil de Golgi. Ensuite, elle est exportée à la membrane
cellulaire, où elle va rester 5 heures en moyenne. Au bout de ces 5 heures,
elle est internalisée par la cellule pour être dégradée dans les endosomes
et les lysosomes.
Les
chercheurs américains ont montré que c’était au cours de cette internalisation
que la protéine changeait de forme et devenait résistante aux protéases.
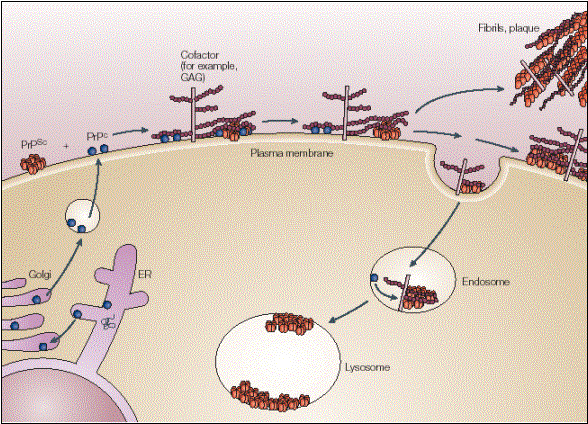
Différences
tridimensionnelles entre PrP-c et PrP-res
Il
y a deux grands types de structure des protéines :
-
Les
hélices alpha qui ressemblent à des escaliers en colimaçon
-
Les
feuillets bêta plissés qui ressemblent à de la tôle ondulée
La
protéine normale a majoritairement des hélices-a, alors que la protéine pathologique a majoritairement
des feuillets ß plissés.
Grâce
à K. Wüthrich, biophysicien de Zurich, on sait à quoi ressemble la protéine
PrP-c (normale).
-
Elle
possède trois hélices a et deux petits
feuillets ß plissés.
-
Il
existe deux zones dans cette protéine :
o
Une
zone hautement structurée, avec les hélices a et les feuillets ß
plissés
o
Une
queue flexible, sans structure prédéfinie.
Aujourd’hui,
on ne connaît pas la structure de la protéine pathologique. Donc on ne sait
pas si l’hypothèse du prion est la bonne. Mais cependant on peut faire des
modèles :
Lorsque
la protéine normale devient pathologique, l’une des trois hélices disparaît
et à la place de cette hélice apparaissent 4 feuillets ß plissés, sans
modification de la séquence primaire de la protéine.
Rôle du
système immunitaire
Pour être infecté par un
prion, il faut un bon système immunitaire. Il existe des souris
manipulées que l’on appelle souris « SCID ». Elles ont une
mutation qui a pour conséquence la suppression de tout le système
immunitaire. On peut leur greffer un système immunitaire qui les rende à
nouveau immunocompétentes.
-
Si
on inocule des souris « SCID » avec un prion, seules 30% tombent
malades, alors que les souris qui ont le même fond génétique mais sans la
mutation « SCID » sont malades dans 100% des cas.
-
Si
on restaure le système immunitaire des souris « SCID » avant de
les inoculer, elles deviennent susceptibles à l’infection par le prion.
Le processus
de la contamination par voie orale
Lors
d’une exposition, le prion traverse la muqueuse digestive, probablement au
niveau des cellules M et le premier site de réplication seront les plaques
de Peyer du tube digestif.
A
partir des plaques de Peyer, transporté certainement par le lymphocyte B,
l’agent va être emmené dans les organes lymphoïdes secondaires (rate,
ganglions lymphatiques).

Dans
les organes lymphoïdes secondaires, il va aller se localiser dans une
catégorie cellulaire, que l’on appelle cellules folliculaires dendritiques
« FDC » (voir schéma). Ces cellules sont dans des centres
germinatifs et ce sont elles qui guident les lymphocytes B pour leur
indiquer le type d’anticorps qu’il va falloir synthétiser. Tout près de ces
cellules dendritiques, il existe des filets nerveux ortho-sympathiques.
L’agent utilise cette proximité, entre la cellule immunocompétente dans
laquelle il peut se multiplier et les filets nerveux, pour entrer dans le système
nerveux périphérique « ENS » et ensuite remonter peut-être par
voie axonale rétrograde ou par infection des cellules de Schwann jusqu’au
ganglion de la racine postérieure. Ensuite l’agent pénètre dans la moelle
et utilise les cordons postérieurs pour se disséminer à l’ensemble du SNC.
5.7. La Maladie de
Creutzfeld Jakob iatrogénique
5.7.1. La
porte d’entrée peut être centrale (SNC)
Le
scénario est toujours le même :
-
Un
patient subit un prélèvement ou est opéré alors qu’il n’a pas encore
déclaré les signes de MCJ et qu’il n’existe pas de test détecteur pour en
faire le dépistage.
-
On
transplante le prélèvement à un autre sujet, ou on utilise les mêmes
instruments de chirurgie avec des procédures de stérilisation d’avant 1995
non aux normes « prion », et on transmet ainsi la maladie.
On
dénombre, ainsi, en France :
-
3
cas après transplantation de cornée
-
7
cas après acte neurochirurgical
-
123
cas après greffe de dure mère. Ceci explique pourquoi les greffes de dure
mère sont interdites en France d’autant qu’il existe des solutions
alternatives.
5.7.2. La voie
d’entrée peut être périphérique
C’est
le cas lors de traitement par à l’hormone de croissance extractive (plus de
130 cas). Cette hormone était préparée à partir d‘hypophyses prélevées dans
les morgues jusqu’en 1985-87. On a dû prélever 1 ou 2 hypophyses chez des
personnes qui étaient infectées, mais qui étaient mortes d’autre chose et
l’on a ainsi contaminé des lots entiers d’hormone, d’autant que les
instruments de manipulations et de recueil n’étaient pas soumis à des
méthodes de désinfection efficace.
5.7.3. Une
observation exceptionnelle…
Une
femme de 67 ans arrive à Lausanne avec un syndrome épileptique particulier
justifiant une exploration avec des électrodes de stéréotaxie en vue de
localiser le point de départ en vue de sa destruction. Tout ce passe bien.
L’infirmière chef de bloc décontamine les électrodes à l’oxyde d’éthylène
sans aucune erreur. Dans les semaines qui suivent, on opère un jeune homme
de 17 ans, une jeune fille de 23 ans pour la même raison et avec les mêmes
électrodes.
La
première patiente, au lieu de s’améliorer, s’aggrave. Le chirurgien. Il
pense non seulement à la MCJ, mais au risque éventuel et il met les
électrodes dans son bureau sous clef. Malheureusement 16 mois et 20 mois
plus tard le jeune homme et la jeune fille meurent de MCJ.
A
ce moment là, il envoie les électrodes, à Béthesda aux USA. Les chercheurs
américains refont sur deux chimpanzés avec les mêmes électrodes l’opération
faite sur les 2 jeunes gens et 20 mois plus tard les 2 chimpanzés meurent.
Deux ans n’altèrent pas l’infectiosité !
La
voie d’incubation conditionne la durée d’incubation et les signes
cliniques :
-
Si
on prend ces 2 jeunes gens, c’est comme si on les avait inoculé par voie intracérébrale, la période d’incubation a été courte de 16 et 20 mois. Cliniquement
ils ont présenté une démence
comme dans la MCJ habituelle.
-
Si
on regarde les enfants qui ont été traité par l’hormone de croissance par voie S.C. ou I.M., ce n’est plus
une contamination par voie intracérébrale, c’est une contamination par voie périphérique, la période d’incubation n’est
plus de 16 à 20 mois , elle est de 5 à 35 ans. Cliniquement n’est plus
une démence mais une grande ataxie
cérébelleuse.
Grâce
aux Inspecteurs régionaux de la santé et aux épidémiologistes de l’INSERM,
on sait que la période de contamination en France a eu lieu entre le 1°
janvier 1984 et le 1° avril ou 1° juin 1985.
968
enfants ont été traités, pendant cette période, 80 sont morts.
|
Année
|
USA
|
RU
|
France
|
|
1996
|
15
|
16
|
39
|
|
2001
|
29
|
30
|
80*
|
(*
80 cas morts, mais il y en a 6 qui sont actuellement malades, et pendant 15
à 20 ans d’autres cas vont apparaître)
5.8. La susceptibilité
génétique
Ces
maladies de Creutzfeld-Jakob iatrogéniques sont à la frontière de la
génétique et des maladies infectieuses. Ceux qui développent la maladie
n’ont-ils pas une génétique particulière ?
On
a recherché les mutations dans le gène de la Protéine P. On n’en a pas
trouvé.
5.8.1. Le
codon 129
On
retrouve un polymorphisme au niveau
du codon 129, totalement silencieux au plan clinique et biologique. A
ce niveau :
-
Peuvent
être codé à ce niveau soit une méthionine soit une valine
-
On
peut être soit
o
homozygote :
méthionine/méthionine, ou valine/valine
o
hétérozygote :
méthionine/valine
-
Dans
la population générale 50 % des gens sont homozygotes, 50 % sont
hétérozygotes
Dans
les MCJ liés à l’hormone de croissance, plus de 90% sont homozygotes au
codon 129. Les Français, les Anglais et les Américains ont le même résultat
dans leur série.
Les études ont montré
que l’homozygotie au codon 129 était le facteur de susceptibilité à toutes
les formes de MCJ y compris les formes sporadiques.
5.8.2. Le
codon 178
On
était surpris de voir qu’une même mutation au codon 178 pouvait entraîner
soit une Insomnie fatale soit une MCJ familiale. Maintenant on sait
pourquoi :
-
Lorsque
le gène qui est muté en 178 a une méthionine en 129 è une Insomnie fatale.
-
Lorsque
le gène qui est muté en 178 a une valine en 129, è une MCJ.
Donc
c’est la mutation qui induit la maladie, mais c’est la structure du gène en
amont de la mutation qui détermine le phénotype clinique.
5.8.3. Quels
sont ces agents infectieux. ?
Aujourd’hui
encore, on ne sait pas.
-
Certains
disent, c’est un agent infectieux, donc il y a un acide nucléique et une
sorte de virus.
-
Pour
d’autres, l’élément majeur c’est la protéine PrP qui est pathologique, car
quand on enlève la PrP pathologique on enlève l’infectiosité, donc l’agent
est la protéine PrP pathologique. C’est l’hypothèse du prion. L’hypothèse
du prion c’est que toute la pathogénicité est contenue dans la structure
tridimensionnelle anormale de la protéine.
5.9. La propagation de
conformation pathologique
La
protéine PrP normale est synthétisée, exprimée à la surface de la cellule
et au bout de 5 heures internalisée pour être dégradée. Le calcul
informatique permet de montrer qu’à partir de la structure de la protéine
normale stable on peut générer une forme normale instable que l’on appelle
PrP*. La demi-vie de cette forme instable est très brève et très rapidement
elle redonne la forme stable.
Si
au moment de la forme instable générée, il y a dans l’environnement une
molécule pathologique, il va y avoir accrochage de la forme normale
instable à la forme pathologique. Cette dimérisation entre la protéine
normale et protéine pathologique fait que la protéine normale change de
forme et adopte la forme de la protéine à laquelle elle s’est accrochée.
C’est la théorie de la propagation de conformation pathologique par
interaction directe protéine/protéine.
6. L’encéphalopathie
spongiforme bovine
6.1. Historique
1°
cas suspect en 1985
1°
cas démontré en 1986.
Entre
fin 1986 et 1988 nos collègues vétérinaires, médecins et scientifiques
britanniques ont tout fait, tout compris et ont fait un travail
remarquable :
-
ils
ont montré que c’était une maladie à prion
-
ils
ont décrit la clinique, l’ont transmise expérimentalement et trouvé
l’origine : des farines de viandes et d’os préparée à partir de
carcasses de ruminants
-
ils
ont proposé la seule mesure de bon sens à leur gouvernement :
l’interdiction des farines de viande et bœuf dans la nourriture des
ruminants. C’est ce que l’on a appelé le «Feed back». Cette mesure
administrative fut introduite en mi-88.
6.2. La maladie
Elle
met 5 ans à incuber. Les effets d’une mesure administrative mi-1988 ne se
voit qu’à partir de 1993. Effectivement à partir de 1993 le nombre de cas
augmente de façon presque exponentielle.
Les
animaux qui sont nés après ce feed back, n’auraient jamais dû développer
d’encéphalopathie bovine spongiforme. Or, il y en a 40 000 que l’on appelle
animaux naïfs, car nés après l’interdiction des farines. Trois hypothèses
peuvent être émises :
-
La
persistance dans l’environnement de l’agent infectieux : on sait que
l’agent est très résistant et peut persister dans l’environnement pendant
des années. La forme actuelle de l’épidémie britannique ne permet pas de
donner à la contamination de l’environnement un rôle majeur pour la
propagation de la maladie.
-
La
transmission mère-veau : elle existe mais à un taux faible (5 % environ)
quand la mère est dans la dernière année d’incubation.
-
La seule explication des
40 000, ce sont des fraudes sur la distribution des farines. Maintenant on le sait
après les enquêtes policières.
6.3. La dangerosité
vis à vis de l’homme
Il
existe trois niveaux de risque : la promiscuité avec les bovins
infectés, la consommation de lait et des produits laitiers, la consommation
de viande et d’abats.
Promiscuité
avec les bovins infectés.
Quatre
agriculteurs britanniques ont développés une MCJ alors qu’ils avaient eu
des vaches folles dans leur troupeau. Cela n’a rien à voir avec
l’encéphalopathie bovine. On peut être quasiment affirmatif sur ce point,
car ils ont l’âge, la clinique, la neuropathologie et les propriétés
biologiques de MCJ sporadiques.
Consommation
de lait et des produits laitiers.
Le
lait n’est pas vecteur d’infectiosité, ne transmet pas la maladie dans
l’état actuel des connaissances quelle que soit l’espèce. Il y a une
maladie humaine, le Kuru, une maladie des papous de Nouvelle-Guinée où
persistait jusqu’à une date récente un cannibalisme avec consommation du
cerveau de la victime. Une étude a porté sur 600 femmes papous qui ont
nourri au sein alors qu’elles étaient en fin d’incubation ou en début de
maladie clinique. Aucun des enfants n’a développé la maladie de leur mère. La transmission par le lait est à
écarter.
La
consommation de viande et d’abats
Le
muscle squelettique ne transmet pas la maladie. Ce sont les abats et
particulièrement le cerveau qui présentent un risque majeur. Le beefsteak
n’est pas vecteur de l’agent, s’il n’est pas contaminé par du tissu nerveux
lors des processus de découpe des viandes.
6.4. L’importance du
problème
|
Etat du nombre de vaches folles en mars 2000
|
|
RU
|
France
|
Eire
|
Suisse
|
Portugal
|
Allemagne
|
Italie
|
|
180
000
|
484
|
800
|
450
|
500
|
6*
|
10
|
(*
chaque cas était signalée comme une bête venue de GB ou de Suisse, jusqu’au
jour où il fut démontré que la bête n’était ni anglaise, ni suisse et
depuis le nombre a augmenté)
Il
n’y a pas de pays actuellement épargné en Europe.
On
peut considérer aujourd’hui qu’il y a eu 180 000 cas de vaches folles au
Royaume Uni et moins de Ζ 000 dans le reste de l’Europe. Donc le
niveau de risque est plus important au Royaume-Uni qu’en dehors.
6.5. Le risque pour
l’homme
L’ampleur du
problème
On
pense que 900 000 bovins infectés sont passés dans la chaîne humaine, et le
pic de l’exposition de l’homme fut en 1988. Pour mémoire, un bovin, compte
tenu des techniques industrielles actuelles, peut se retrouver dans 200 000
assiettes, ceci pour donner l’idée de la dispersion potentielle que peut
avoir l’agent. S’il y a un risque pour l’homme, c’est cela qu’il faudra
gérer.
Un modèle
explicatif
Pour
estimer le risque il faut tenir compte du lieu de localisation de l’agent
infectieux. Malheureusement la distribution de l’agent infectieux varie (en
dehors du système nerveux) d’une espèce à l’autre et d’une souche de prion
à l’autre.
La
vache folle n’a du prion que dans son cerveau quand elle est à la phase
clinique. Mais le problème n’est pas la vache folle qui est détectée par le
vétérinaire à l’abattoir et qui est alors mise hors du circuit alimentaire.
Ce qui est important c’est où se situe l’agent infectieux chez la vache en
incubation de la maladie.
Les
britanniques ont fait l’expérience suivante : ils ont pris 40 veaux,
ils les ont exposé à l’agent bovin par voie orale et tous les 2 ou 3 mois
ils ont tué 2 veaux et ils ont re-inoculé les tissus de ces tissus à des
souris pour tracer l’agent infectieux. Ils ont vu que
-
Pendant
les 4 premiers mois on ne détecte pas l’agent
-
A
partir du 6ème mois on détecte l’agent dans la partie distale de l’intestin
grêle. Cette positivité de l’iléon distal reste présente jusqu’à la mort de
l’animal.
-
A
partir du 30-34ème mois, l’agent devient détectable dans le ganglion
rachidien
-
Du
36ème mois, dans le SNC
-
A
38-40 mois la vache a des signes cliniques.
Ceci
explique pourquoi les intestins des bovins sont interdits à la
consommation.
La
transmissibilité
L’autre
façon d’estimer le risque, c’est de voir si cet agent est transmissible à
d’autres espèces facilement. Parmi toutes les espèces testées par voie
intracérébrale, il n’y a guère que le hamster et le poulet qui soient
totalement résistants. Tous les autres y sont sensibles.
Dans
la MCJ sporadique, on n’a jamais trouvé l’agent infectieux dans les
ganglions lymphatiques.
En
revanche, dans les 27 cas de MCJ nouvelle variante (nv-MJC) qui ont été
étudiés, on a trouvé l’agent dans 27 cas/27 dans la rate, dans les
ganglions, dans les amygdales. C’est dire que l’agent est à haut titre en
dehors du SNC dans cette nouvelle forme de MCJ. Aujourd’hui, dans la nv
MCJ, le diagnostic peut être fait par l’examen d’une biopsie de l’amygdale
(aspect spécifique).
Deux
patients britanniques ont été opérés de l’appendicite 11 mois et 3 mois 1/2
avant que les signes cliniques de nv-MCJ n’apparaissent. Les neurologues
ont récupéré les pièces opératoires dont l’examen a montré la présence de
la protéine P pathologique. Tous les actes chirurgicaux deviennent des
actes à risque et pas seulement les actes neurochirurgicaux !
Les
deux paramètres que l’on attend sont :
-
Le
nombre de cas à venir. Cet élément va déterminer le risque pour la
population.
-
Y
a-t-il une infectiosité ou non dans le sang ?
7. Conclusions
On
est en face d’une maladie émergente !

|