|
EPU95 – Montmorency
Neurologie
 Mise à jour du 11 Mai 2007 Mise à jour du 11 Mai 2007 

Sclérose en
Plaques en 2003
Dr T. de Broucker
Chef de service de Neurologie (Hôpital
Delafontaine - Saint Denis)
Séance du 3 avril 2003
La dernière décennie (1993 à 2003) a été marquée, en ce
qui concerne la Sclérose en Plaques (SEP), par d’importants progrès
thérapeutiques, diagnostiques et méthodologiques. Il importe de faire le
point de ce qu’il convient de faire aujourd’hui quand on soupçonne une SEP
et lors de sa prise en charge.
1. QUELQUES CAS
CLINIQUES DE SEP
La SEP n’est pas une maladie d’aspect et d’évolution
monomorphe. Au contraire, elle se présente sous des aspects fort variables
qui permettent de décrire des histoires différentes : banale –
difficile – catastrophique – agressive – inquiétante - mono-symptomatique -
progressive démentielle – pseudo-tumorale – bénigne – primaire progressive
lente sans poussée.
1.1. Une histoire banale
Une jeune femme de 22 ans, secrétaire, sans antécédent
personnel ou familial.
En 2000, elle présente un engourdissement du membre
supérieur gauche qui régressera et à l’examen clinique un trouble sensitif
objectif très discret.
-
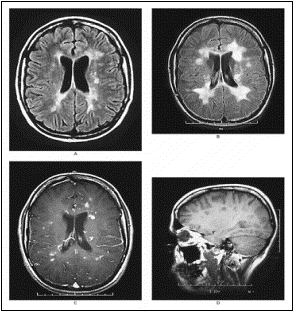
L’IRM montre des zones d’hypersignal (ZHS) en T2
sustentoriels dont l’une très nette assez évocatrice d’une zone de
démyélinisation de SEP. En T1, il n’y a pas de prise de Gadolinium (Gd -).
-
A la PL, le liquide est normal avec la présence de
bande oligoclonale (BO +) à l’électrophorèse.
Devant la forte suspicion d‘épisode de démyélinisation,
elle reçoit des flash IV de méthylprednisolone (Solumédrol®) pendant 3
jours.
La symptomatologie s’améliore pour disparaître.
Trois mois plus tard, elle est revue : clinique
normale avec EDSS à 0 et IRM de contrôle avec régression des ZHS.
Elle n’est revue qu’en 2003 pour des dysesthésies de localisation
similaire du moignon de l’épaule (avec sensation d’une épaule gonflée) et
des troubles sensitifs objectifs discrets de la main. A l’IRM une
modification modérée et apparition de nouvelles ZHS en T2 et toujours Gd –
en T1.
Reprise du traitement par flash de méthylprednisolone et
mise en route d’un traitement par interféron.
1.2. Un cas difficile non sur le plan diagnostic
mais sur la conduite à tenir
Une J.F. étudiante, qui a 20 ans en 1996 lorsqu’on la
découvre.
Mais elle a déjà présenté en 93 un épisode de
paresthésie de l’hémicorps gauche ayant duré 3 semaines à 1 mois sans
qu’elle ait consulté ni eu de bilan ni de diagnostic.
De même en janvier 95, elle présenta une diplopie
horizontale pendant quelques semaines. Elle a reçu une vaccination contre
l’hépatite B quelques semaines auparavant pour s’inscrire à une école
paramédicale.
En 1996, elle présente un déficit moteur du membre
inférieur droit. A l’IRM des ZHS + , la PL est positive avec BO+. Le bilan
systémique est négatif ce qui permet de poser le dg de SEP devant
l’histoire avec poussées successives, l’IRM et la PL.
Le traitement repose là encore sur le Méthylprednisolone
et l’on débute le traitement par interféron ß1b (début de l’interféron dans
la SEP) qu’elle tolère mal avec des accès pseudo-grippaux persistants ce
qui nécessite son arrêt en juin 97.
En février 1998, nouvelle poussée de troubles sensitifs
de l’hémicorps droit. Elle est mise sous un autre interféron ß1a jusqu’en
novembre 1998 où elle présente des hallucinations au cours des injections.
Le traitement est arrêté sans savoir si les hallucinations étaient réelles
ou liées au psychisme particulier de la patiente.
De 98 à 2002, elle fait de nouvelles poussées sensitives
ou motrices de l’hémicorps droit, mais la réalité de chaque poussée est
discutable car on n’arrive pas à mettre en évidence des éléments
d’aggravation. Elle a cependant une aggravation de l’IRM avec une atrophie
cérébrale. Son EDSS est stable à 3 et elle travaille normalement. Elle est
mise sous Imurel® 150 mg par jour avec une très bonne tolérance.
En décembre 2002, elle fait une nouvelle poussée
sensitive du membre inférieur.
Cette histoire est une histoire difficile, car cette
J.F. a un psychisme particulier avec un comportement revendicatif vis-à-vis
de la vaccination VHB, ce qui rend délicat l’interprétation de la
symptomatologie subjective ainsi que la conduite thérapeutique car si les
flash de Méthylprednisolone sont de courte durée, ils ne sont justifiés
qu’à bon escient.
1.3. Une histoire de SEP catastrophique
Mlle S. a 20 ans en 1993 et présente un premier épisode
de diplopie et un syndrome cérébelleux.
Le diagnostic de SEP est rapidement fait et le
traitement est symptomatique par le méthylprednisolone. A l’IRM de 93, il y
a des anomalies du tronc cérébral en T2, et des prises de contraste
multiples au Gd+ au niveau encéphalique (corticale et corps calleux).
Elle ne récupère pas correctement après la première
poussée. L’évolution est faite de rémissions partielles et surtout
l’évolutivité de son déficit l’oblige, à peine 5 ans après le début de la
maladie, à vivre en fauteuil roulant. Elle n’a pas reçu de traitements de
fond car il n’y en avait pas de disponible sur le marché à cette période-là
(96), et lorsqu’ils sont apparus elle avait un handicap trop important qui
ne correspondait plus à l’AMM.
Cette patiente avait un caractère difficile, des liens
avec la drogue et un mode de vie particulier.
De 93 à 2001, l’IRM montre de nouvelles lésions
multiples importantes et l’aggravation de l’atrophie en particulier au
niveau du corps calleux (facilement mesurable à cet endroit, et constituant
un bon marqueur de l’atrophie cérébrale).
En 2000, elle a une grossesse qui sera menée à terme.
Dès l’accouchement, 1 mois plus tard elle présente une
nouvelle poussée avec majoration des troubles déficitaires et cérébelleux
des 4 membres. Traitement de méthylprednisolone. En fév 2001, apparaissent des troubles de la déglutition et
une insuffisance respiratoire aiguë nécessitant une prise en charge en
réanimation (traitement symptomatique et antibiotique). A l’arrêt des
antibiotiques, alors que le poumon est nettoyé, on n’arrive pas à la sevrer
de la machine. A l’IRM , le bulbe n’est plus qu’un ensemble de plaques
également disséminées sur l’ensemble du tronc cérébral avec une prise de
Gd+ partout. Son trouble d’insuffisance respiratoire aiguë était une
poussée de la SEP et non simplement liée à des fausses-routes. Après 10
jours de flash de Solumédrol® , l’amélioration des troubles ventilatoires
permet le passage en service de neurologie mais dans un état grabataire et
avec une amnésie majeure puisqu’elle ne se rappelle plus qu’elle a un
enfant né 3 mois plus tôt. Son EDSS
est à 10.
Elle retourne chez elle où son mari et la mère de
celui-ci s’occupent d’elle. En avril 2001, 8 ans après le début elle décède
brusquement.
1.4. Une histoire de SEP agressive
Cette J.F a 14 ans en 1996 et présente une
symptomatologie de névrite optique rétrobulbaire probable (baisse de la
vision) puisqu’elle n’a pas été examinée à ce moment-là, symptomatologie
qui fut régressive.
Quelques mois plus tard en septembre 96, apparaissent
une diplopie verticale, une marche pseudo-ébrieuse, une algodynie du gros
orteil qui sont régressives en quelques semaines.
En février 97, récidive de la même symptomatologie
encore régressive. Et en avril 97 elle est envoyée par son médecin traitant
pour dysesthésies des deux mains et engourdissement du tronc à un médecin
interniste qui la confie au neurologue. L’IRM est impressionnante avec de
nombreuses ZHS et prise de Gd+ . On est en droit de craindre une évolution
aussi calamiteuse que le cas précédent.
Le diagnostic de SEP est porté. Elle reçoit des flash de
Solumédrol® et la symptomatologie rentre dans l’ordre de façon complète.
Elle est mise sous interféron ß1b pendant quelques mois. Un syndrome
dépressif très sévère et une intolérance hépatique font arrêter le
traitement en décembre 97.
En 98 elle fait une 5ème poussée. Elle est mise sous
interféron ß1a (Avonex®) de mars 98 à août 99. Entre temps malgré le
traitement immuno-modulateur, des poussées surviennent ( août 98 - mars 99
– mai 99 – août 99).
A cette 9ème poussée, elle est mise sous Imurel® d’août
99 à décembre 99 et pendant cette période elle a encore 3 poussées. On est
devant une forme extrêmement évolutive mais laissant très peu de séquelles
après chaque poussée. Elle est considérée comme une forme agressive et mise
sous chimiothérapie par mitroxantrone (Novantrone®) 12 mg /m2 par voie IV
tous les 3 mois.
De décembre 99 à novembre 2000 pas de poussée. A l’IRM
de 2000 il existe une atrophie du corps calleux plus marquée qu’en 1997.
En novembre 2000, elle refait un trouble visuel, et en
décembre 2000 nouvelle poussée avec des rires immotivés. Ce tableau montre
le polymorphisme de ces poussées puisqu’à chaque fois ce sont des symptômes
différents et parfois difficiles à rattacher à un événement neurologique.
Elle reçoit encore des flash de Solumédrol® et on essaie de raccourcir le
délai entre 2 chimiothérapie (6 semaines au lieu de 3 mois).
En octobre 2001, c’est la 13ème et dernière cure de
Novantrone® (dose maximale totale autorisée) et en mars 2003 elle n’a pas
fait de nouvelle poussée. Son EDSS est à 2,5. Vie socio-professionnelle
normale.
1.5. Une autre histoire de SEP agressive
inquiétante
Une femme de 25 ans qui a un antécédent familial de SEP
chez une tante. En avril 2002, elle présente une hypersomnie, un déficit de
l’hémicorps droit, une hémianopsie latérale homonyme gauche. A l’IRM des
lésions multiples sus-tentorielles qui prennent le Gd+. Elle est mise sous
corticothérapie orale (ce qui n’est pas à faire).
En mai 2002, c’est-à-dire 1 mois plus tard elle présente
une névrite optique, un syndrome cérébelleux bilatéral. Hospitalisée en
neurologie, le bilan de maladie systémique est négatif et le LCR montre l’existence
de bandes oligoclonales permettant le diagnostic de SEP. Elle reçoit 3
flash de Solumédrol®.
La symptomatologie régresse totalement.
En août 2002 en vacances dans le sud de la France, elle
présente un déficit moteur du membre inférieur droit, un syndrome cinétique
cérébelleux des 2 membres supérieurs. Elle reçoit 3 flash de Solumédrol®.
Trois semaines plus tard, elle est réhospitalisée en urgence pour un état
de mal épileptique. A l’IRM découverte d’une grosse lésion qui prend le Gd+
avec des lésions importantes sustentorielles. En réanimation, elle reçoit
des antiépileptiques, 3 flash de Solumédrol® et mise en route de
l’interféron ß1a (Rebif® un peu plus dosé que l’Avonex®) 3 fois par
semaine.
En mars 2003 à l’IRM les prises de contrastes ont disparu,
les grosses lésions se sont nettement améliorées et elle n’a pas eu de
nouvelles manifestations cliniques. L’EDSS est à 1 et elle mène une vie
normale.
1.6. Une forme monosymptomatique de SEP
Cet homme à l’âge de 29 ans présente en avril 2001 une myélopathie
subaiguë pseudo-tumorale qui l’amène à consulter. Il a une lésion
médullaire qui prend le Gd+ d’où une biopsie le 1° mai. L’histologie est en
faveur d’une affection démyélinisante. Le geste a été décidé sans doute
devant une suspicion d’abcès. En juin 2001, compte tenu du résultat
d’ana-path un bilan cérébral est effectué qui montre des ZHS multiples
sustentoriels. Le LCR est positif avec des bandes oligoclonales à
l’électrophorèse. Il garde des séquelles sous forme de déficit
sensitivo-moteur du membre supérieur droit qui est apparu dans les suites
de l’intervention.
En octobre 2002, il vient consulter à l’hôpital
Delafontaine pour avoir un autre avis, car il était virulent à l’égard du
geste chirurgical qu’il avait subi. L’IRM montre les mêmes images que l’on
avait lors de l’examen précédent auxquelles s’adjoignent de nouvelles
lésions ce qui permettait de porter le diagnostic de SEP et autorise de
débuter un traitement immuno-modulateur par interféron.
1.7. Une histoire de SEP progressive démentielle
Mr. D. est un
patient qui en 1993 fait une poussée unique se traduisant par un syndrome
cérébelleux statique une dysarthrie, un rire et pleurer spasmodique, une
incontinence urinaire. L’ensemble du bilan est positif pour le diagnostic
de SEP et l’IRM de l’époque montre déjà une atrophie cérébrale ce qui
laisse supposer qu’il a dû faire auparavant des poussées non reconnues. Il
s’agissait d’un mécanicien auto.
En 1994, il a une aggravation progressive avec un
syndrome pyramidal et un trouble intellectuel ainsi que des manifestations
psychotiques à type de délire systématisé. Son EDSS est à 7, traduisant une
évolution rapidement progressive à partir d’une 1ère poussée en 1993.
Il a été transféré en milieu psychiatrique et il est
décédé quelques années plus tard.
1.8. Une forme pseudo tumorale
A la mi février 2003, une patiente présente un syndrome
grippal. Le 24 février apparaissent une paralysie de la main gauche et une
hémiparésie de l’hémicorps gauche. A l’IRM il existe une aspect tumoral ou
pseudo-tumoral sans effet de masse. Dans le service d’accueil, le
diagnostic est celui d’accident vasculaire cérébral. Elle sera transférée
en neurologie où le LCR est d’aspect inflammatoire (25 éléments). Les
potentiels évoqués visuels et auditifs étaient normaux. L’IRM montre en fait à côté de la grosse
lésion d’autres petites anomalies de haut signal qui pourraient être en
rapport avec des lésions démyélinisantes.
On ne peut affirmer qu’il s’agit d’une SEP, car, même si
à l’électrophorèse du LCR on découvrait des bandes oligoclonales, il
pourrait s’agir d’une encéphalomyélite aiguë disséminée à poussée unique
qui ne récidive pas.
1.9. Une forme bénigne
Cette patiente a débuté sa maladie en 1989 avec des
troubles sensitifs aux 2 membres inférieurs avec un syndrome pyramidal
réflexe, une impériosité mictionnelle. Elle a eu un traitement qui à
l’époque reposait sur 7 flash un jour sur deux d’1,5 gr de Solumédrol® (2
semaines d’hospitalisation).
En 1992, elle présente une névrite optique rétrobulbaire
gauche. Elle reçoit 5 « flashes » de Solumédrol®.
De 1992 à 2002 elle a 2 grossesses qui évoluent bien.
En 2002, elle a un discret syndrome pyramidal, une
impériosité mictionnelle. A l’IRM il existe des prises de contraste au
Gd+. Instauration d’un traitement
immuno-modulateur par copolymer.
1.10. Une forme primaire progressive lente sans
poussée.
Mme D. a 56 ans et depuis 1982 elle a une
paraparésie progressive (raideur des 2 membres inférieurs à la marche. Le
diagnostic de SEP est posé à la Salpétrière.
4
En 1988 une IRM est pratiquée montrant quelques
anomalies et une atrophie dorsale que l’on voit dans ces formes.
4
En 1992, c’est-à-dire 10 ans après le début, elle
utilise une canne.
4
En 1997, elle arrête son activité professionnelle.
4
En 1999, elle utilise 2 cannes et souvent un
fauteuil roulant.
C’est une forme qui n’a jamais fait de poussée.
Au regard de ces différentes formes de SEP on voit qu’il
s’agit d’une maladie touchant des personnes jeunes dont le pronostic est
lourd et que même les formes dites bénignes ne le sont pas. On peut
souhaiter à un patient en début de maladie qu’il se trouve dans le clan des
malades ayant une affection à évolution lente et surtout que les progrès
thérapeutiques obtenus depuis 10 ans vont encore s’améliorer.
2. EPIDEMIOLOGIE
EN 2003 DE LA S.E.P.
La prévalence de la sclérose en plaque est de 25 à 60
pour 100 000 habitants en France.
Le sex-ratio est de 0,5 c’est-à-dire un homme pour 2
femmes.
La fréquence du début de la maladie est fonction de
l’âge :
4
85 % des cas entre 16 à 40 ans
4
10 % des cas > 40 ans
4
5 % des cas à un âge < 16 ans
2.1. Une particularité de la SEP est d’être
géographiquement dépendante :
Il y a plus de SEP au Nord de l’hémisphère Nord et au
Sud de l’hémisphère Sud. La zone équatoriale étant relativement épargnée.
Une autre remarque, a été soulevé par J.F. Bach dans un
article récent concernant des maladies auto-immunes comme la SEP mais aussi
la maladie de Crohn, le diabète type 1 ou l’asthme. Ces maladies sont en
augmentation constante dans les zones où les maladies infectieuses
(tuberculose, oreillons, rougeole, hépatite A,…) diminuent.
J.F. Bach proposent devant cette constatation, deux
orientations de recherche :
-
Est-ce que les infections multiples
permettent-elles l’acquisition d’une certaine tolérance vis-à-vis d’auto-antigène
comme ceux par exemple de la myéline pour la sclérose en plaques ?
comme si l’absence de cette stimulation infectieuse entraînerait pour
l’organisme la capacité de développer une auto-agressivité immunitaire.
-
Est-ce que les infections nombreuses survenant
pendant l’enfance permettent-elles l’intervention de cytokines d’origine
lymphocytaire dans la modulation des populations des cellules
immuno-compétentes ? de telle sorte que l’absence de production de
cytokines favoriserait le développement de clones auto-réactifs chez
l’individu qui n’aura pas été suffisamment infecté dans l’enfance.
Le risque géographique, s’acquiert dans l’enfance :
-
Si l’on vit son enfance jusque vers 15 ans dans un
pays à faible risque et que l’on migre après dans un pays à fort risque, on
garde le risque de l’enfance.
-
Par contre, si on migre tôt dans son enfance d’un
pays à faible risque vers un pays à haut risque, on acquiert le risque du
pays qui l’accueille.
Ces constatations montrent le rôle que joue
l’environnement dans ce type de pathologie.
2.2. S.E.P. et vaccinations
L’étude de l’équipe de Confavreux de Lyon à partir d’une
base de données très importante sur la SEP a permis de montrer de façon
claire l’absence d’influence des vaccinations sur la survenue d’une poussée
de SEP déjà connue.
On discute encore en France du rôle de la vaccination
VHB dans le déclenchement d’une SEP.
2.3. SEP et grossesse
La grossesse n’influe pas sur l’évolution de la maladie.
Confavreux a étudié l’influence de la grossesse sur les poussées de la SEP,
(étude de femmes présentant une grossesse au cours de leur affection et qui
ont été particulièrement surveillées pendant les 3 trimestres de la
grossesse et les 3 mois du post-partum). Il a observé :
-
une décroissance importante du nombre de poussées
durant le 3ème trimestre de la grossesse,
-
et par contre une ré-augmentation du nombre de
poussées après l’accouchement.
-
mais le nombre total de poussées pendant cette
année de surveillance est le même que celui d’une année sans grossesse.
De cette étude, deux questions ont été soulevées :
-
Ne faudrait-il pas induire un traitement
immuno-modulateur après l’accouchement pour éviter la poussée du
post-partum ?
-
Une hormonothérapie à base d’œsto-progestatif dans
le traitement de fond ne serait-elle pas bénéfique ? Des essais
cliniques sont en cours.
2.4. SEP et génétique
A côté du rôle de l’environnement que l’on connaît mal,
celui d’une prédisposition génétique a pu être démontré.
-
Chez les jumeaux homozygotes, en moyenne dans
toutes les études, la concordance que les deux fassent la maladie est de 25
%. Alors que chez les jumeaux hétérozygotes le risque n’est que de 5 % qui
est à peu prés le même que celui qui existe entre 2 membres d’une même
fratrie (4 %).
-
De même entre enfant et parent de sujets atteints,
le risque est de 2,75 %.
-
Ce risque génétique qui a été étudié est
indépendant de l’environnement.
-
Il existe également un risque lié à la race, en
particulier les Caucasiens chez lesquels le risque est plus élevé que chez
les noirs ou les asiatiques.
3. LE DIAGNOSTIC
EN 2003
3.1. Le diagnostic
Il repose sur l’association de 2 critères :
-
la dissémination spatiale de la maladie
-
la dissémination temporelle
Les outils de diagnostic sont : l’anamnèse, la
sémiologie, l’imagerie, la biologie et l’électophysiologie :
L’imagerie
-
Jusqu’à ces dernières années, l’imagerie n’était
pas un critère de diagnostic mais plutôt un argument du diagnostic
différentiel
-
L’imagerie est rentrée tout récemment (2002) dans
les critères utilisables pour le diagnostic positif de SEP.
L’électrophysiologie a perdu beaucoup de son intérêt
depuis l’apparition de l’IRM :
-
Les potentiels évoqués (auditifs, visuels,
sensitifs),
-
L’exploration urodynamique, qui montre dans la SEP
avec troubles sphinctériens un tracé très suggestif de la maladie.
3.2. Deux catégories de critères
Les critères de Poser (1983)
Ce sont des critères anciens :
-
la dissémination spatiale,
-
la dissémination temporelle
-
et le caractère inflammatoire du LCR
Ces critères permettent de décrire 3 catégories de
SEP :
-
cliniquement probable,
-
biologiquement probable,
-
probable cliniquement et biologiquement.
Si le bilan ne permet pas de rentrer dans une de ses
catégories, ce n’est pas une SEP au jour de la consultation.
 Ces critères de Poser sont
toujours utilisés, mais ils sont actuellement en concurrence avec les
critères de MacDonald. Ces critères de Poser sont
toujours utilisés, mais ils sont actuellement en concurrence avec les
critères de MacDonald.
Les critères de Mac Donald (2001)
Ils utilisent l’imagerie IRM qui permet de décrire 2
critères : le critère spatial/inflammatoire et le critère temporel
-
SEP certaine (critère spatial / inflammatoire du
LCR + critère de dissémination temporelle)
-
SEP possible lorsque un de ces deux critères est
absent.
-
sinon ce n’est pas une SEP
Les critères de l’IRM de Barkhof (1997) Les radiologues
doivent maintenant avoir un œil calculateur quand il voit une SEP pour
savoir si l’on rentre ou non dans les critères IRM.
Dans une étude importante de 1997, Barkhof est parti de
syndrome cliniquement isolé (première poussée de SEP) et a analysé les IRM
successives ; il suivait les patients cliniquement et faisait une
corrélations entre les observations cliniques et l’IRM, ce qui lui a permis
de suivre le devenir du patient en terme de SEP définie.
On peut lors d’une poussée initiale prévoir dans 85% des
cas une poussée dans les années à venir, lorsque 3 des 4 critères suivant
sont réunis :
-
Une lésion qui prend le gadolinium en T1 ou 9 zones
d’hypersignal (ZHS) en T2,
-
Au moins 1 lésion sous-tentorielle,
-
Au moins 1 lésion juxta-corticale
-
Au moins 4 lésions périventriculaires.
Le scanner est beaucoup moins puissant et ne montre pas
l’ensemble des anomalies que l’on trouve à l’IRM.
A partir de l’IRM on a pu définir le critère de
dissémination temporelle. Le radiologue doit pour évaluer ce critère
disposer d’un examen antérieur. Le critère de dissémination temporelle
repose sur :
-
L’existence de lésions rehaussées par le Gd+, 3
mois ou plus après une poussée, car la prise de contraste que l’on voit au
cours d’une poussée à l’IRM disparaît en fait en moins de 2 semaines le
plus souvent. Si on en voit 3 mois après, cela traduit une nouvelle poussée
même si elle est infraclinique.
-
L’apparition de nouvelles ZHS au moins 3 mois après
une IRM précédente.
Les études de ces dernières années concernant l’IRM ont
permis de montrer que le nombre de poussées découvertes à l’IRM était 5 à
10 fois supérieur par rapport aux poussées avec critères cliniques.
3.4. Le diagnostic différentiel se pose
avec :
-
Des maladies qui sont très proche de la SEP :
-
Maladie de Devic qui touche l’œil et la moelle
épinière,
-
La sclérose concentrique de Balo qui est une SEP
pseudotumorale
-
La maladie de Schilder qui est une sclérose en
plaques de l’enfant,
-
L’ADEM qui est une encéphalomyélite démyélinisante
unique qui peut être extrêmement grave mais qui ne se reproduit pas
-
Des maladies inflammatoires systémiques sont
nombreuses qu’il faut absolument éliminer : BBS, Behcet, Gougerot,
lupus érythémateux aigu disséminé,
-
Des maladies infectieuses : syphilis, maladie
de Lyme, infection par HTLV1.
-
Les maladies vasculaires : le syndrome des
antiphospholipides, le Cadasile qui est une maladie génétique des petits
vaisseaux
-
Les tumeurs,
-
les malformations vasculaires ou de la charnière
vertébro-occipitale
-
Les maladies génétiques ou métaboliques dans les
formes progressives primaires (dg difficile car l’IRM peut être normal).
4. ÉVOLUTION ET
PRONOSTIC
4.1. Formes cliniques
Il y a 5 formes cliniques qui ont été détaillées
initialement dans les tableaux de présentation :
4
La forme à poussées
§
Sans séquelles
§
Avec des séquelles
§
Progressives secondaire
4
Les formes sans poussée mais progressive secondaire
avec poussées
4
La forme progressive d’emblée, beaucoup plus rare,
avec ou sans poussée
La poussée traduit un phénomène inflammatoire alors que
la progression traduit un phénomène dégénératif.
4.2. Définition de la poussée
« La poussée
est une manifestation neurologique focale nouvelle ou une aggravation
franche durant plus de 24 heures », hors contexte infectieux.
4
L’installation des symptômes se fait en général en
quelques jours
4
La durée est en général de quelques semaines,
4
La régression est le plus souvent complète dans les
formes à poussées
4
Dans les formes à poussées avec séquelles la
régression est partielle
4
La NORB (40 % des modes de révélation) se traduit
par des douleurs de l’œil notamment à la mobilisation oculaire volontaire
du sujet ou lors d’essai de mobilisation par l’examinateur, une baisse de
l’acuité visuelle, un scotome central, un FO normal.
La poussée survient hors contexte infectieux. La fièvre
exacerbe ou refait apparaître des symptômes car elle entraîne un déficit de
conduction des fibres démyélinisées. Un bain chaud peut entraîner le même
phénomène.
Une poussée doit être séparée d’un mois au minimum de la
précédente.
4.3. Diagnostic différentiel d’une poussée
4
Le phénomène d’Uthof qui survient à l’effort (un
peu comme le fait la fièvre), concernant surtout la névrite optique,
l’effort entraînant alors une baisse de la vision.
4
L’épilepsie partielle avec une phase post-critique
qui peut mimer une poussée,
4
Les pseudo-poussées chez des patients anxieux ayant
l’impression et la crainte qu’il y a quelque chose qui revient.
4.4. Le handicap
Le repérage de l’handicap au cours de l’évolution doit
être systématique lors de chaque consultation à plusieurs niveaux :
moteur, sensitif, visuel, sphinctérien, cognitif, épilepsie, fatigue chronique,
douleurs.
Ce repérage permet d’établir un score, l’EDSS (expanded
disability status scale) reproductible d’une consultation à l’autre et
transmissible d’un médecin à l’autre.
4
Les fonctions motrices : la fonction
pyramidale (déficit, raideur), les fonctions cérébelleuses (l’ataxie en
particulier et de très loin le plus invalidant)
4
Les fonctions du tronc cérébral : de 0 à 3
(nystagmus, paralysie oculo-motrice) de 4 à 5 (troubles bulbaires ou
pseudo-bulbaires)
4
Les fonctions sensitives
4
Les fonctions visuelles (principalement la baisse
de l’acuité liée à la NO et pouvant régresser, mais aussi à l’ataxie du
regard)
4
Les fonctions sphinctériennes intestinales mais
surtout vésicales et les problèmes d’incontinence
4
Les troubles sexuels trop souvent oubliés d’apprécier
4
Les troubles cognitifs souvent précoces
4
L’épilepsie peu fréquente, souvent tardive, pouvant
être la traduction d’une poussée
4
La fatigue chronique pouvant entraîner chez des
sujets qui n’ont aucun déficit moteur une invalidité réelle et difficile à
comprendre par l’entourage familial et/ou professionnel.
4
Les douleurs fréquentes et variées, devant être
appréciées et traitées en fonction des mécanismes en cause
(désafférentation, ou due à la paralysie).
5. LE PRONOSTIC
Confavreux a étudié 1844 patients qui se répartissent en
gros en 85 % de formes rémittentes à rechute et en15 % de formes
progressives primaires. Il a regardé par le biais de l’EDSS, un score
ambulatoire qui est noté à :
4
« 4 » : périmètre de marche limité à
moins de 500 mètres
4
« 6 » : une canne devient nécessaire
à la marche
4
« 6,5 » : deux cannes sont
nécessaires
4 « 7 » :
le fauteuil roulant est le moyen de se déplacer.
Il a regardé sur l’importante population de son étude
suivie de façon linéaire avec toujours l’EDSS depuis 1980, ce que devenaient
les patients en fonction de leur forme clinique. Le temps d’évolution à
partir du diagnostic est le suivant :
|
Temps pour arriver
|
Formes
rémittentes à rechutes
|
Formes progressives primaires
|
|
Grade
« 4 »
|
11 ans
|
Lors du diagnostic (formes spinales)
|
|
|
Grade
« 6 »
|
23 ans
|
7 ans
|
|
|
Grade
« 7 »
|
33 ans
|
13 ans
|
|
|
|
|
|
|
Il y a, en gros, 20 ans d’écart pour arriver au grade 7
entre la forme à rechute et la forme progressive primaire.
Si l’on prend les patients à partir du grade 4 et que
l’on regarde le temps qu’ils mettent pour arriver au grade 6 ou 7, on
s’aperçoit que le délai est le même dans les deux formes : pour
arriver au grade 6 dans la population à rechutes il faut 5,7 ans, et dans
celle à évolution progressive primaire il faut 5,4ans.
La différence principale entre les deux formes c’est une
différence d’âge de début : en gros il y a une différence de 20 ans
dans l’âge de début suivant la forme : rémittente à rechutes début
précoce, primaire progressive début beaucoup plus tardif.
On a l’impression que l’évolution de la SEP est âge
dépendant et que le processus dégénératif commence probablement au même
moment dans l’âge de l’individu. On peut déterminer des éléments de bon et
mauvais pronostic.
|
Eléments de bon pronostic
|
Eléments de mauvais pronostic
|
|
-
Etre une femme
-
Avoir un début rémittent
-
une récupération complète à chaque poussée
-
de longs intervalles inter-poussée
-
une fréquence basse des poussées au début
-
un délai élevé avant d’atteindre l’EDSS 3
-
un âge jeune
|
-
Etre un homme
-
Avoir un début polysymptomatique et moteur
-
notamment cérébelleux
-
une récupération incomplète
-
un bref intervalle entre les poussées
-
des fréquences élevées des poussées au début
-
un délai bref avant l’invalidité
-
l’âge plus élevé
|
|
|
|
|
|
Si on regarde l’âge jeune ou l’âge élevé du début de la
maladie, on s’aperçoit que le sujet jeune lorsqu’il va arriver à un âge
plus élevé il va tomber dans le mauvais pronostic.
Ces éléments de pronostic n’ont de valeur que pour aller
du grade 0 au grade 4 de l’EDSS.
6. TRAITEMENT
6.1. Physiopathologie.
Il s’agit d’une
atteinte inflammatoire démyélinisante du système nerveux. Il existe
également une atteinte axonale.
La perte de myéline au niveau de la gaine laisse l’axone
à nu qui peut être lésé. Un axone sans gaine perd son rôle de
conductibilité de l’influx nerveux, mais la gaine peut être revitalisée.
Il est probable que, à côté de la démyélinisation de la
gaine, l’atteinte axonale soit présente immédiatement dès les premières
phases de la maladie et non pas secondairement lorsque la régression est
incomplète, d’où l’idée du phénomène dégénératif associé au phénomène
inflammatoire.
Pour le mécanisme du phénomène inflammatoire, de
nombreuses hypothèses sont actuellement soulevées.
Le mécanisme de la dégénérescence axonale dans la mesure
où il n’est pas que secondaire reste plus mystérieux.
Le schéma actuellement retenu pour comprendre
l’évolution de toute SEP (Confavreux) : « poussées et atteinte
axonale progressive sont concomitantes mais cliniquement décalées liées à
l’âge et la durée d’évolution de la maladie.
6.2. Les cibles thérapeutiques
Lutter contre le phénomène inflammatoire :
-
La corticothérapie : méthylprednisolone
-
L’immuno modulation : essentiellement les
interféron ß avec leur problème de tolérance (la voie orale pour le moment
hypothétique mais utilisable déjà dans la PCE), les immunoglobulines
polyvalentes.
-
L’immunosuppression : Imurel®,
cyclophosphamide, mitoxantrone
-
Les autres voies pour agir sur l’inflammation et
notamment l’inhibition des molécules d’adhésion
La réparation myélinique :
-
Si à chaque poussée on peut réparer la myéline
comme avant, on peut imaginer obtenir une action de réparation quasi
complète sinon complète et améliorer l’évolutivité e la maladie.
La neuroprotection
-
Elle s’adresse au phénomène dégénératif.
-
Pour l’instant, les recherches sur ce sujet n’ont
pas abouti.
6.3. Traitement des poussées
La corticothérapie par voie intraveineuse (et non la voie orale) à
fortes doses
Elle est actuellement le traitement de la poussée. Le protocole nécessite une
hospitalisation à cause des incidents qui sont survenus lors de la
perfusion du Solumédrol® dans d’autres pathologies.
Il utilise :
-
le méthylprednisolone (Solumédrol®) 1gr par jour
pendant trois jours de suite,
-
puis de la prednisone pendant onze jours (cette 2ème
partie du protocole est parfois oubliée)
-
La corticothérapie diminue l’intensité et la durée
des manifestations neurologiques mais n’a pas d’effet prouvé sur le
handicap à moyen et long terme. Elle n’a aucun intérêt en dehors des
poussées.
6.4. Traitement de fond des formes rémittentes à
rechute
6.4.1. Les interférons ß.
L’efficacité
Ces médicaments sont utiles aux patients. Leur
efficacité est similaire, quel que soit le produit utilisé, sur les
différentes formes cliniques entraînant une diminution de 30 % des poussées
cliniques et une diminution des poussées visibles à l’IRM de 80 à 90 % ce
qui est très important.
4
Les
interférons ß1a : l’Avonex® (1 injection par semaine 30 mg) et le
Rebif® (3 injections par semaine) qui sont des interférons naturels
4
L’interféron
ß1b dont le premier qui est sorti est le Bétaféron® (250 mg tous les
deux jours).
La tolérance.
L’interféron ß peut entraîner :
-
-Un état pseudo grippal : L’interféron ß étant
la molécule que l’on secrète lors d’une grippe, l’injection peut donner un
syndrome de type grippal pendant quelques heures voire 24 h (fièvre,
frissons, douleurs) lors de l’injection, pouvant être invalidant, ou modéré
mais aussi s’estomper au fil des injections et ne nécessiter que la prise
de paracétamol.
-
Problèmes de tolérance cutanée
-
Le syndrome dépressif n’est pas un état secondaire,
mais il est une contre indication de leur emploi.
-
L’intolérance hématologique ou hépatique est rare.
6.4.2. Les autres traitements de fond :
L’acétate de glatiramer (Copaxone™)
C’est un analogue de la myéline, dont on ne connaît pas
son mode d’action. Il est un petit
peu moins actif que les interférons mais de tolérance excellente.
L’Imurel™
C’est un immunosuppresseur est utilisé depuis 20 ans par
les Lyonnais et qui, malgré l’absence de preuve d’efficacité, continuent de
l’utiliser.
Dans leur grande cohorte, ils se sont aperçus que dans
certains cas il y avait un arrêt total des poussées chez des patients
sélectionnés
Les autres options possibles
Des données très récentes semblent montrer que le cyclophosphamide (Endoxan®)
pourrait avoir un intérêt dans certaines formes de SEP agressives
Les immunoglobulines
polyvalentes : un essai encourageant.
6.5. Traitement des
formes secondairement progressives à rechute
6.5.1. Le contexte…
Jusqu’à ces récentes années, il n’y avait pas de
traitement de fond de ces formes.
L’effet bénéfique
de l’interféron ß1b a été démontré dans un essai thérapeutique qui
convainc les Européens. Par contre deux autres essais d’Amérique du Nord
sont négatifs.
Dans les formes secondairement progressives, la mitoxantrone a montré son efficacité
avec maintien de l'effet plus d'un an après la fin de l'essai.
La mitoxantrone est une anthracycline qui bloque la
polymérase.
-
La toxicité cardiaque est dose dépendante et il ne
faut absolument pas dépasser la dose de 140 mg au total.
-
Sa toxicité hématologique n’est pas nulle. Et sur
l’ensemble des SEP traitées dans le monde (autour de 1500), il y a eu 3 cas
de leucémie. Donc grande prudence dans son emploi.
-
La dose totale ne doit pas dépasser 140mgr (20mgr
tous les 3 mois).
On ne sait pas s’il faut prendre le relais par un
immunomodulateur. Cette décision est laissée au clinicien avec l’avis du
patient.
6.5.2. Les traitements symptomatiques
Ils sont d’autant plus justifiés qu’il est reconnu que
plus l’handicap est lourd et plus courte est la consultation de neurologie,
ce qui traduit le désintérêt du clinicien vis-à-vis de ces sujets alors
qu’ils ont besoin de traitements symptomatiques adaptés nécessitant un
travail en équipe.
|
Fréquence dans une
consultation de SEP
|
|
Très
fréquents
|
Moins fréquents
|
|
Troubles
sphinctériens : 80 %
Spasticité :
77 %
Fatigue :
69 %
Troubles
de la marche : 66%
Déficit
du membre inférieur : 64 %
Trouble
du transit : 50%
Douleurs :
49 %
|
Trouble
de la déglutition : 25 %
Trouble
sexuel : 12 %
Dysarthrie
< 10%
Trouble
de l’humeur < 10%
|
Il faudrait avoir une check-list à chaque
consultation :
-
Pour interroger correctement le patient
-
Pour pouvoir envisager les moyens à mettre en œuvre
-
Pour réduire ou atténuer le handicap :
o
Physiothérapie, orthophonie
o
Soins à domicile
o
Prise en charge sociale et psychologique (annonce
du dg.)
Le rôle des associations de malades et d’Internet sont
loin d’être négligeables.
Il n’y a pas
de prise en charge standard de la SEP du fait de l’imprévisibilité de
l’évolution.

|